2020年是极不平凡的一年,面对突如其来的新冠肺炎疫情,国家药品监督管理局药品审评中心(以下简称药审中心)在国家药品监督管理局(以下简称国家药监局)的坚强领导下,认真学习贯彻习近平总书记重要讲话和重要指示批示**,闻令而动、尽锐出战,坚持人民至上、生命至上,超常规建立“早期介入、持续跟踪、主动服务、研审联动”全天候应急审评审批工作机制,加速推动新冠病毒**和新冠肺炎治疗药物研发上市,充分发挥技术审评对疫情防控的科技支撑作用;主动服务于药监系统工作大局,紧紧围绕落实党中央国务院审评审批制度改革、贯彻《药品管理法》《**管理法》《药品注册管理办法》、推动审评体系和审评能力现代化,统筹推进疫情防控和依法依规科学审评工作,不断提高审评质量和效率,不断加快新药研发上市步伐,为疫情防控和满足临床急需提供有效药物保障、为医药产业高质量发展提供有力促进作用,保障了人民群众用药安全有效可及,药品审评事业得到新发展、迈上新台阶、开创新局面。
一、药品注册申请审评审批情况
(一)总体完成情况
1.全年审评审批完成情况
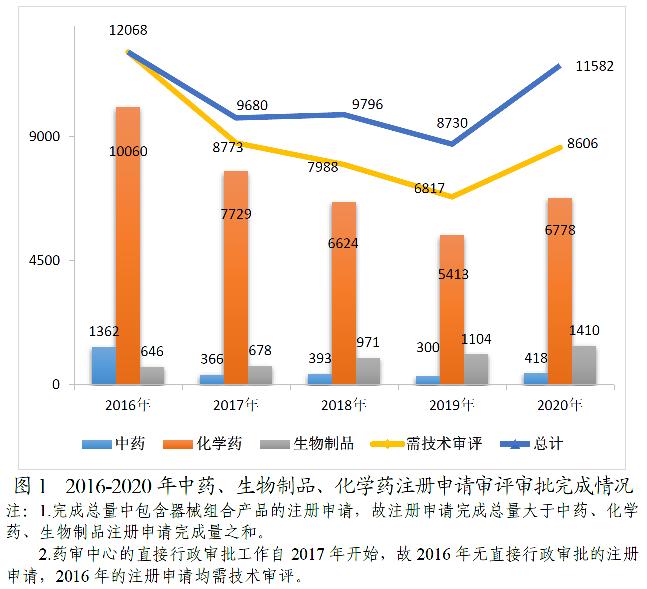
2020年,根据《药品注册管理办法》(国家市场监督管理总局令第27号)、《国家药监局关于实施<药品注册管理办法>有关事宜的公告》(2020年第46号,以下简称46号公告)及《药品注册管理办法》相关配套文件,药审中心完成中药(包括民族药,下同)、化学药、生物制品各类注册申请审评审批共11582件(含器械组合产品4件,以受理号计,下同),较2019年增长32.67%(如无说明,以注册申请件数计,下同)。其中,完成需技术审评的注册申请8606件(含5674件需药审中心技术审评和行政审批注册申请),较2019年增长26.24%;完成直接行政审批(无需技术审评,下同)的注册申请2972件。2020年底正在审评审批和等待审评审批的注册申请已由2015年9月高峰时的近22000件降至4882件(不含完成技术审评因申报资料缺陷等待申请人回复补充资料的注册申请)。
完成8606件需技术审评的药品注册申请中,化学药注册申请为6778件,较2019年增长25.22%;中药注册申请418件,较2019年增长39.33%;生物制品注册申请1410件,较2019年增长27.72%;化学药注册申请约占全部技术审评完成量的78.76%。2016-2020年中药、生物制品、化学药注册申请审评审批完成情况详见图1。
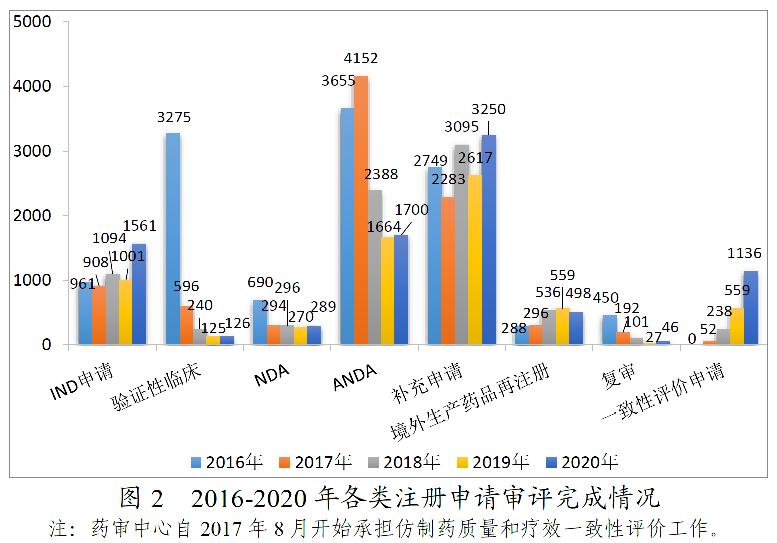
药审中心完成需技术审评的8606件注册申请中,完成新药临床试验(IND)申请审评1561件,较2019年增长55.94%;完成新药上市申请(NDA)审评289件,完成仿制药上市申请(ANDA)审评1700件;完成仿制药质量和疗效一致性评价(以下简称一致性评价)申请(以补充申请途径申报)1136件,较2019年增长103.22%;完成补充申请技术审评3250件,较2019年增长24.19%。2016-2020年各类注册申请审评完成情况详见图2。
3.审评通过情况
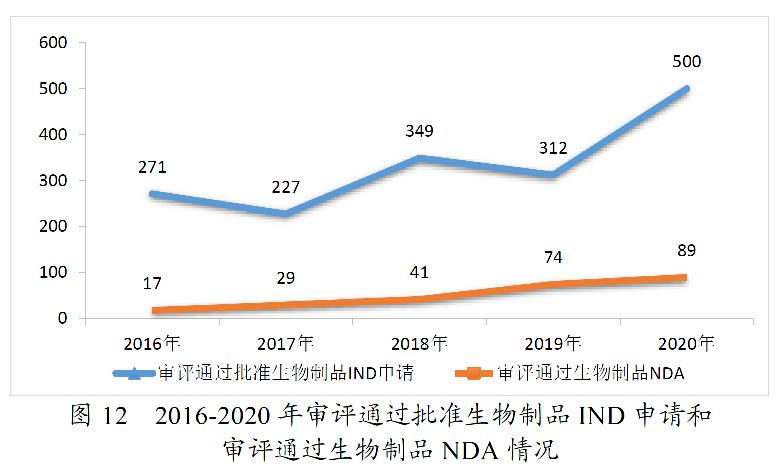
2020年,药审中心审评通过批准IND申请1435件,较2019年增长54.97%;审评通过NDA 208件,较2019年增长26.83%;审评通过ANDA 918件;审评通过批准一致性评价申请577件,较2019年增长121.92%。
药审中心审评通过创新药NDA 20个品种,审评通过境外生产原研药品NDA 72个品种(含新增适应症品种),具体品种详见附件1、2。
4.审结注册申请任务按时限完成情况
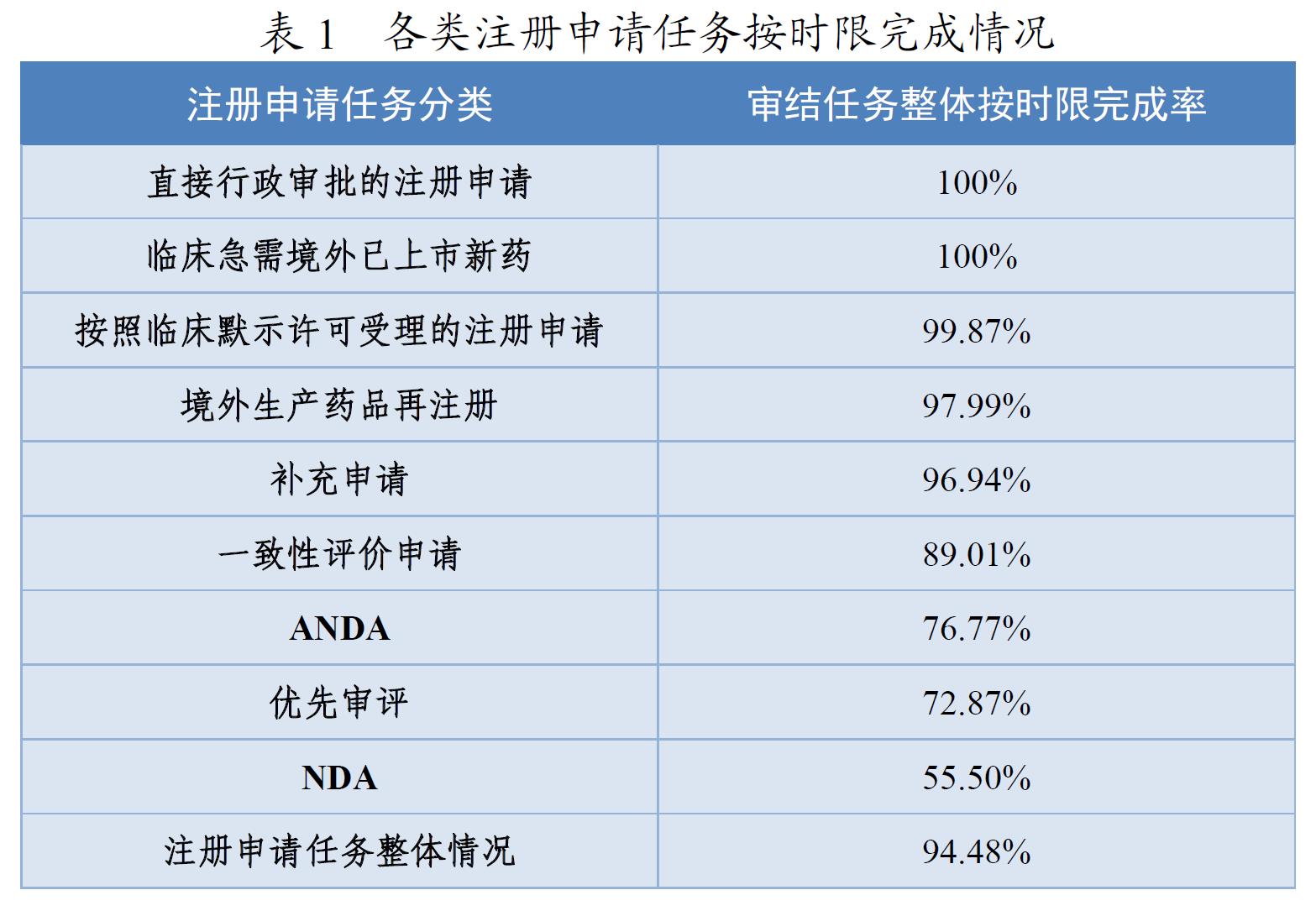
2020年,药审中心持续优化审评流程,严格审评时限管理,加强项目督导,加快审评速度,整体审评任务和重点序列审评任务按时限完成率均取得显著提升。全年审结注册申请任务整体按时限完成率为94.48%,其中临床急需境外已上市新药注册申请审结任务整体按时限完成率为100%,按默示许可受理注册申请的审结任务整体按时限完成率为99.87%,直接行政审批的注册申请100%在法定的20个工作日内完成,且审批平均用时11.8个工作日。各类注册申请任务按时限完成情况详见表1。
2020年的NDA年度整体按时限完成率已经有了很大的提升,例如:NDA按时限完成率在2020年12月突破80%,提升至87.5%;ANDA按时限完成率在2020年12月突破90%,达到93.85%;纳入优先审评程序的注册申请按时限完成率在2020年10-12月的月度按时限完成率连续达到90%以上,取得历史性突破。
(二)中药注册申请审评完成情况
1.总体情况
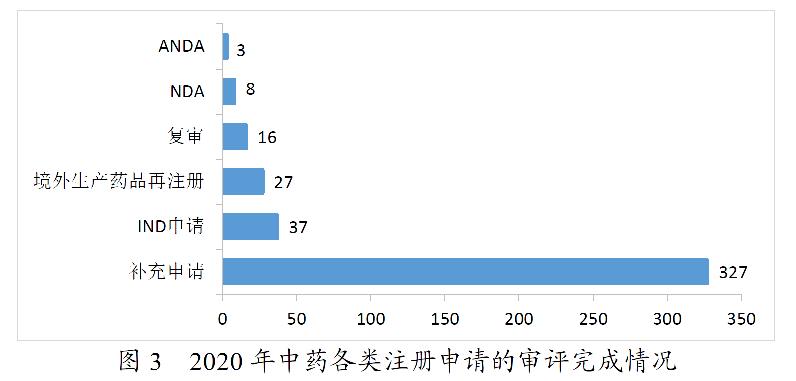
2020年,药审中心完成审评的中药注册申请418件。其中,完成IND申请37件,完成NDA 8件,完成ANDA 3件。2020年中药各类注册申请的审评完成情况详见图3。
2.审评通过情况
药审中心审评通过批准中药IND申请28件,审评通过中药NDA 4件(连花清咳片、筋骨止痛凝胶、桑枝总生物碱片及桑枝总生物碱)。2020年中药各类注册申请审评完成的具体情况详见表2,2016-2020年审评通过批准中药IND申请和审评通过中药NDA情况详见图4。
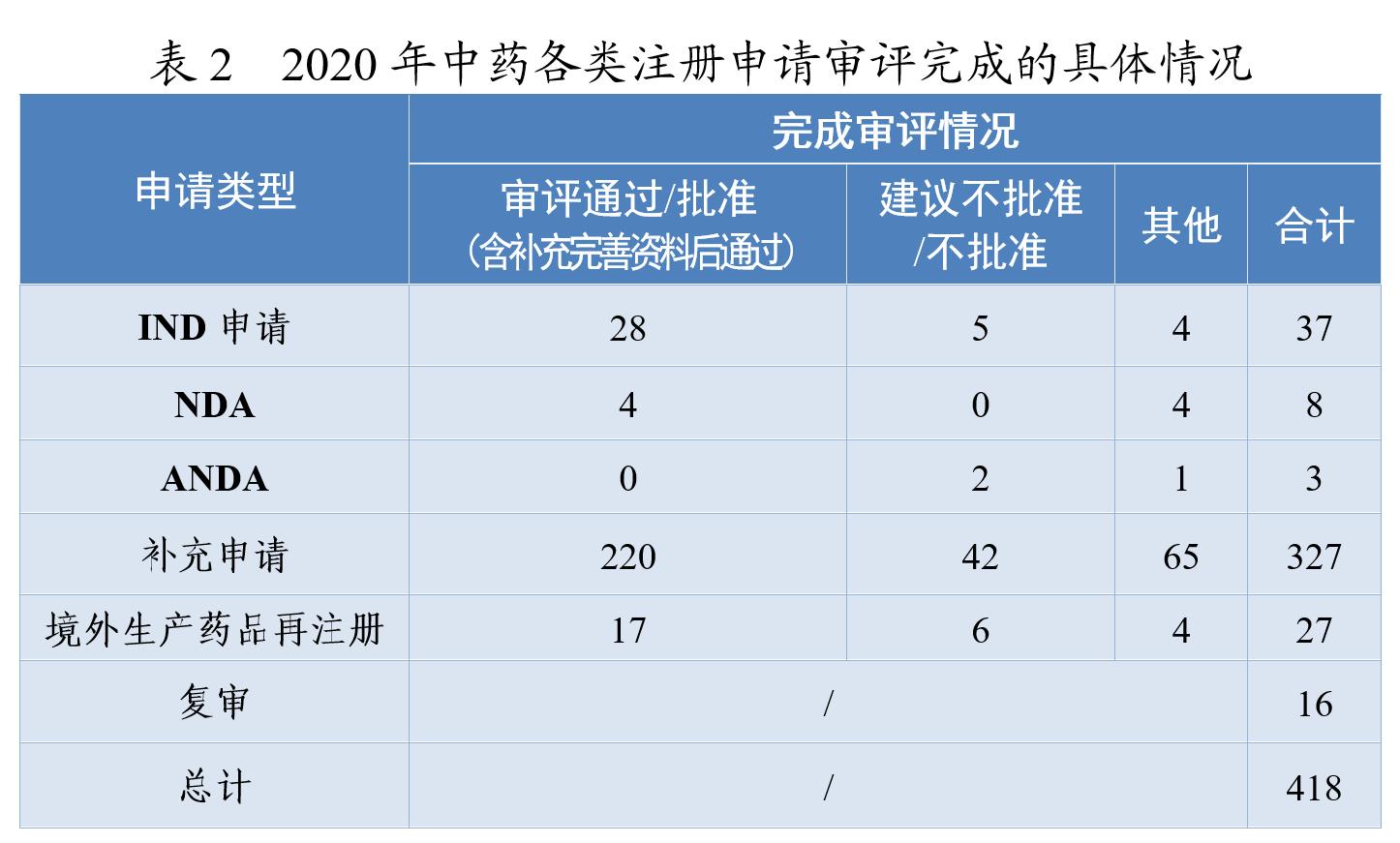
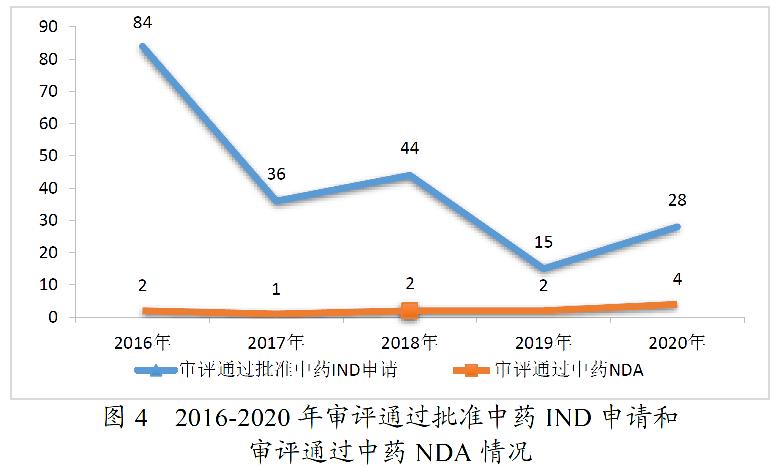
药审中心审评通过批准的中药IND申请28件,涉及10个适应症领域。其中,呼吸7件、骨科4件、消化4件,共占53.57%,2020年审评通过批准的中药IND申请适应症分布详见图5。
(三)化学药注册申请审评完成情况
1.总体情况
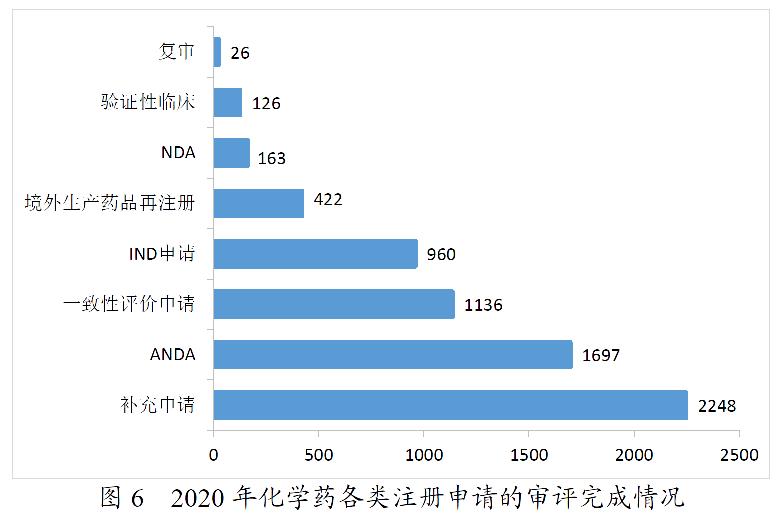
2020年,药审中心完成审评的化学药注册申请6778件。其中,完成化学药临床申请(IND申请和验证性临床)共1086件,较2019年增长45.58%;完成化学药NDA 163件;完成化学药ANDA 1697件;完成一致性评价申请1136件,较2019年增长103.22%;完成化学药补充申请2248件,较2019年增长23.72%。2020年化学药各类注册申请的审评完成情况详见图6。
2.审评通过情况
药审中心完成审评的化学药注册申请中,审评通过批准IND申请907件,较2019年增长51.42%;审评通过NDA 115件,较2019年增长30.68%;审评通过ANDA 918件,较2019年增长15.33%。2020年化学药各类注册申请审评完成的具体情况详见表3。
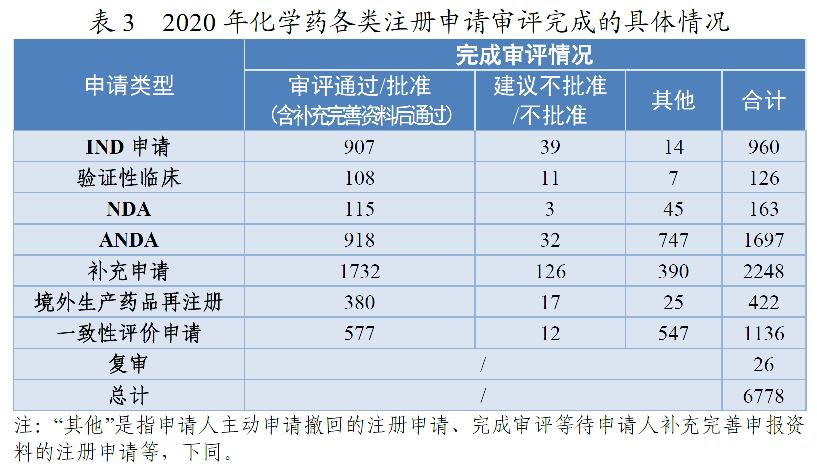
药审中心完成审评的化学药IND申请960件,审评通过批准IND申请907件。其中,1类创新化学药IND申请694件(298个品种),较2019年增长40.77%,品种数较2019年增长57.67%。2016-2020年审评通过批准化学药IND申请、1类创新化学药IND申请情况详见图7。
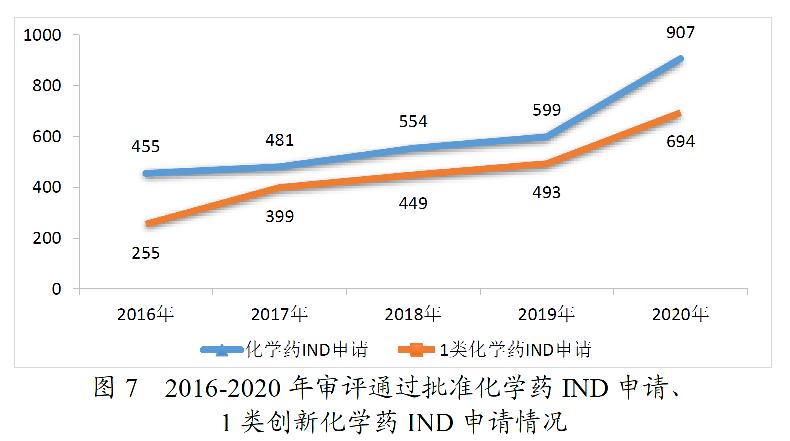
药审中心审评通过批准IND申请的694件1类创新化学药中,抗肿瘤药物、抗感染药物、循环系统疾病药物、内分泌系统药物、消化系统疾病药物和风湿性疾病及免疫药物较多,占全部创新药临床试验批准数量的80.69%。2020年审评通过批准的1类创新化学药IND申请适应症分布详见图8。
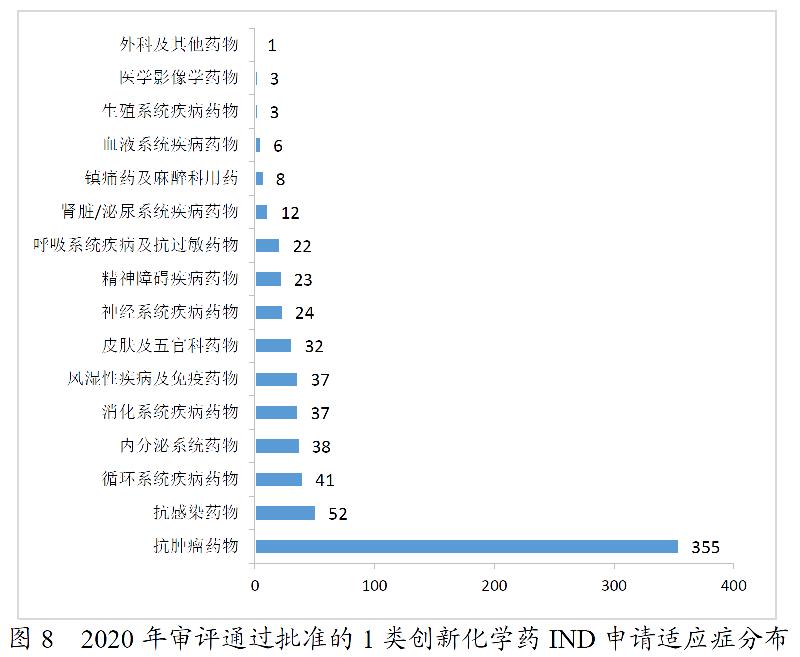
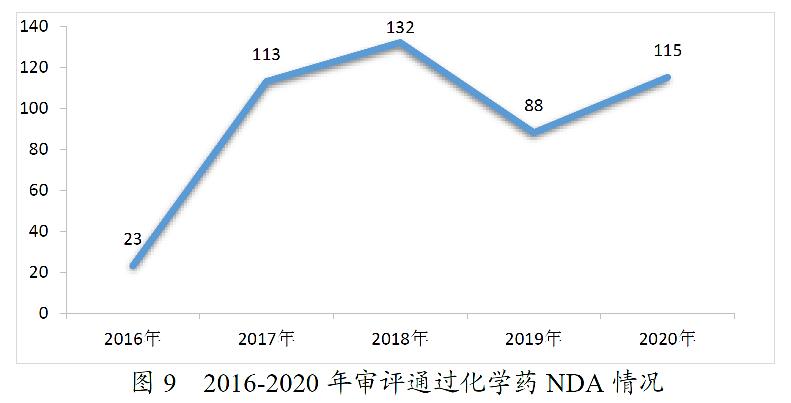
药审中心完成审评的化学药NDA共163件。其中,审评通过化药NDA 115件,审评通过1类创新化学药NDA 14个品种。2016-2020年审评通过化学药NDA情况详见图9。
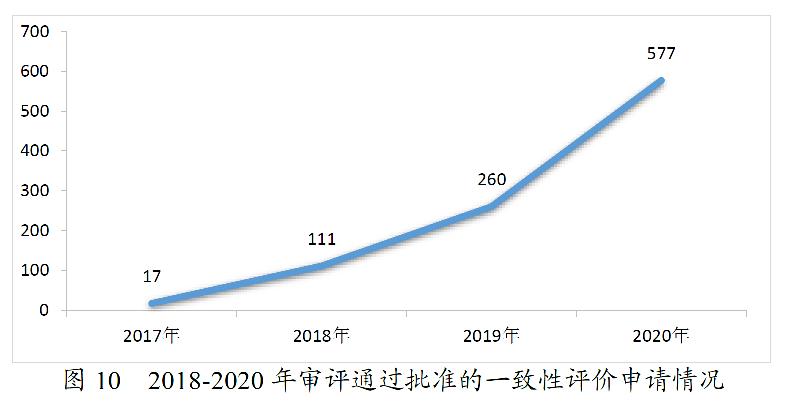
药审中心完成审评的一致性评价申请共1136件,审评通过577件。其中,审评通过批准口服固体制剂一致性评价456件,审评通过批准注射剂一致性评价申请121件,具体品种详见附件3。2018-2020年审评通过批准的一致性评价申请情况详见图10。
(四)生物制品注册申请审评完成情况
1.总体情况
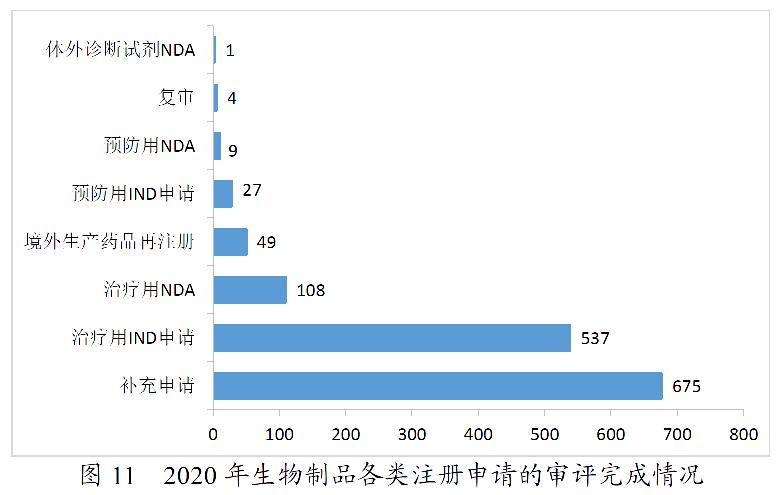
2020年,药审中心完成审评的生物制品注册申请共1410件。其中,完成预防用生物制品IND申请(预防用IND申请)27件,完成治疗用生物制品IND申请(治疗用IND申请)537件,较2019年增长58.88%;完成预防用生物制品NDA(预防用NDA)9件,完成治疗用生物制品NDA(治疗用NDA)108件,完成体外诊断试剂NDA(体外诊断NDA)1件。2020年生物制品各类注册申请的审评完成情况详见图11。
2.审评通过情况
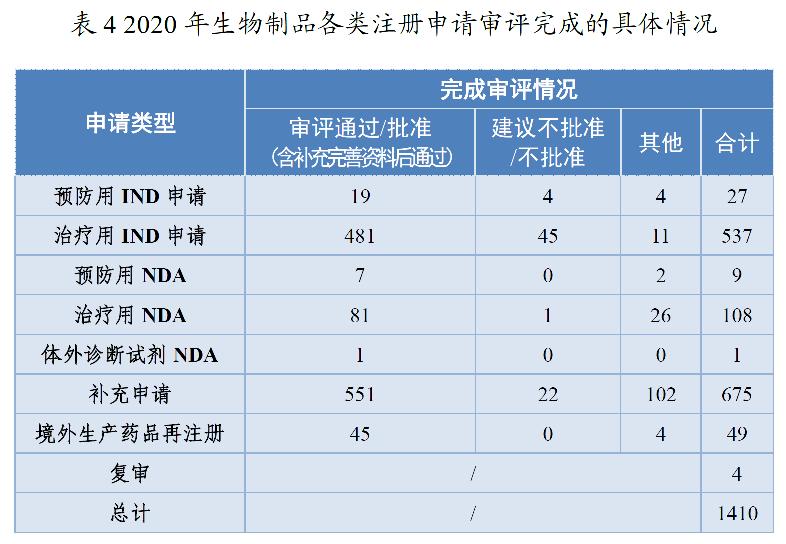
药审中心审评通过批准生物制品IND申请500件,较2019年增长60.26%。其中,预防用IND申请19件;治疗用IND申请481件,较2019年增长63.61%。审评通过生物制品NDA 89件,较2019年增长20.27%。其中,预防用NDA 7件;治疗用NDA 81件(制剂77件),较2019年增长19.12%;体外诊断NDA 1件。2020年生物制品各类注册申请审评完成的具体情况详见表4,2016-2020年审评通过批准生物制品IND申请和审评通过NDA情况详见图12。
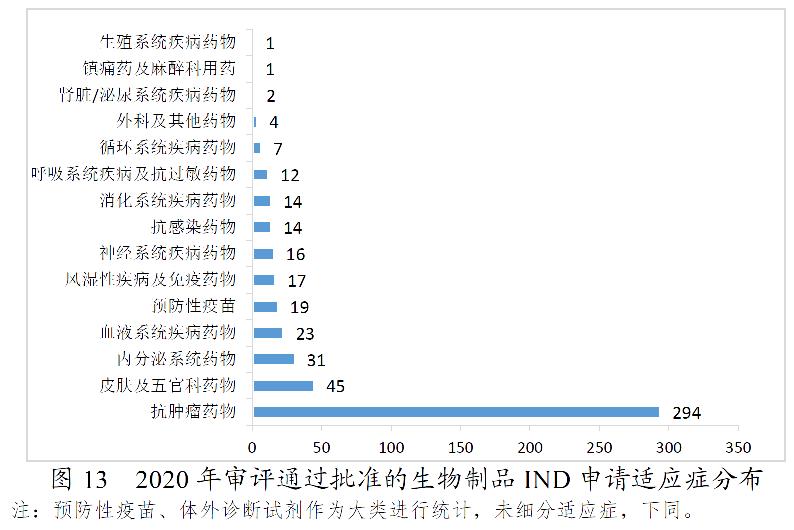
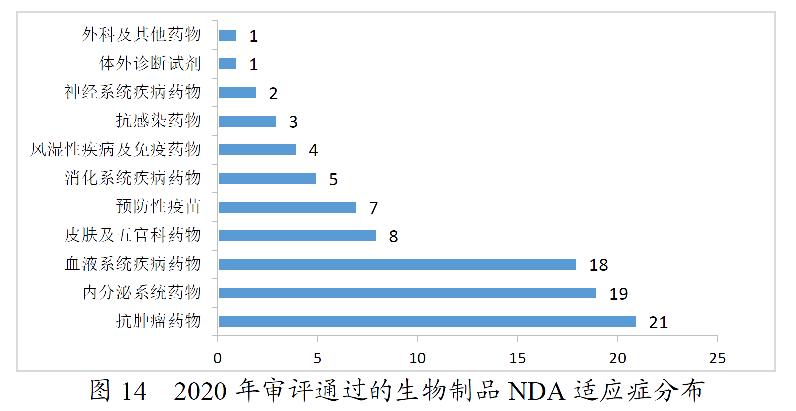
药审中心审评通过批准生物制品IND申请500件,2020年审评通过批准的生物制品IND申请适应症分布详见图13。药审中心审评通过生物制品NDA 89件,2020年审评通过的生物制品NDA适应症分布详见图14。
(五)行政审批注册申请完成情况
1.总体情况
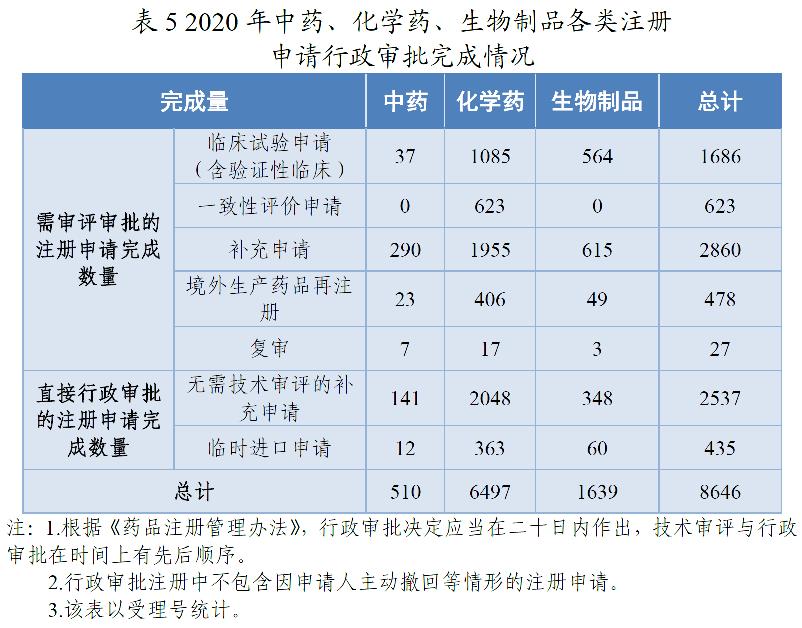
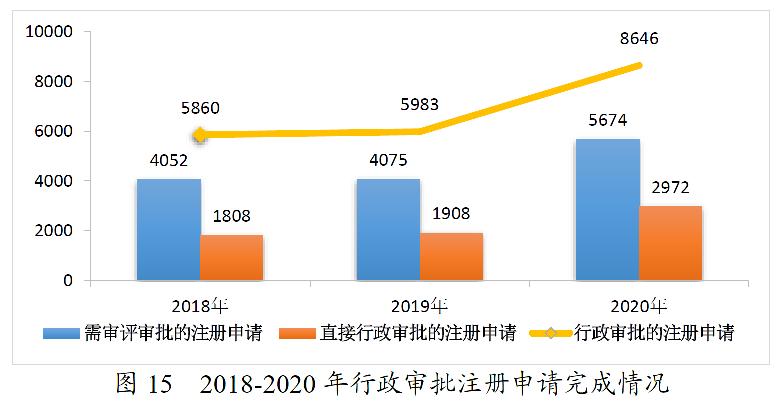
2020年,药审中心完成中药、化学药、生物制品各类注册申请行政审批共8646件,较2019年增长44.51%。其中,完成审评审批的注册申请(临床试验申请、一致性评价申请、补充申请、境外生产药品再注册及复审)5674件,较2019年增长39.24%;完成直接行政审批的注册申请(无需技术审评的补充申请、临时进口申请)2972件,较2019年增长55.77%。2020年中药、化学药、生物制品各类注册申请行政审批完成情况见表5。2018-2020年行政审批注册申请完成情况详见图15。
2.需审评审批的注册申请完成情况
药审中心完成的需审评审批的5674件注册申请中,临床试验申请1686件(含验证性临床),较2019年增长50.00%;一致性评价申请623件,较2019年增长80.58%;补充申请2860件,较2019年增长34.46%;境外生产药品再注册478件、复审27件。
3.直接行政审批的注册申请完成情况
药审中心完成直接行政审批的2972件注册申请中,按注册申请类型划分,补充申请2537件、临时进口申请435件。按药品类型划分,中药153件、化学药2411件、生物制品408件。
(六)注册申请不批准的主要原因及存在的问题
2020年,中药、化学药、生物制品各类药品注册申请因申报资料无法证明药品安全性、有效性或者质量可控性,以及未能按期提交补充资料等情形,导致审评结论为建议不批准的共367件。通过系统梳理上述注册申请不批准原因,从新药、仿制药等不同注册分类角度分析药品注册申请在研发和申报过程中存在的主要问题包括:
1.新药申请
IND申请和研发中存在的问题主要有:正式申报前未开展沟通交流;开发立题依据不足,成药性存在严重缺陷;申报资料不足以支持开展药物临床试验或者不能保障临床受试者安全。具体表现包括:未沟通交流致使申报后发现研究信息严重缺项,无法在时限内完成补充研究;已有的研究结果提示药效作用弱,**大,临床获益和风险比值不合理;临床开发定位违背临床诊疗、用药的基本原则;已有的药学、临床前研究不符合临床试验要求;临床试验方案整体设计严重缺陷,风险控制措施不足;联合用药的非临床研究数据不充分;联合**中单苗的数据不充分和/或免疫程序不一致。
NDA研发和申报中存在的问题主要有:研究质量控制和管理存在缺陷,导致已有的研究结果不能证明药品安全性、有效性和质量可控性;违反合规性要求。具体表现包括:关键临床研究设计存在重大缺陷,无法得出客观、有力的有效性、安全性证据;药学研究存在严重缺陷,无法证明产品的质量可控性;各开发阶段的研究受试样品不一致;注册核查中发现临床试验数据存在真实性问题。
2.仿制药申请
仿制药一致性评价申请和上市申请中存在的问题主要有:仿制药研发立题不合理;申报资料无法证明仿制药与参比制剂(被仿制药品)的质量一致性。具体表现包括:仿制药的参比制剂已撤市,且已有更新换代安全性更好的产品满足临床需求;样品复核检验不符合规定或分析方法存在严重缺陷;人体生物等效性试验结果表明不等效;样品稳定性研究结果、原料药起始物料选择等不符合仿制药上市技术要求;仿制药未按规定使用具有合法来源的原料药。
3.补充申请
补充申请研究和申报中存在的问题主要有:申请资料未能充分说明变更的科学性和合理性,不足以支持变更事项;已有的研究结果不能保证变更后产品的安全性、有效性和质量可控性。具体表现包括:变更引起药用物质基础发生重大改变;药品说明书修改申请不符合说明书撰写的技术要求;用于支持变更的文献资料存在偏倚,或者临床安全性和有效性数据不充分。
4.其他
其他药品注册申请在研发和申报中存在的问题主要有:生物类似药开发缺少相似性比较数据,药学比对研究中参照药选择存在缺陷;生物类似药临床前研究结果不足以支持其开展临床试验;天然药物的研究资料不符合国际多中心临床试验或我国天然药物评价基本技术要求。
(七)药品加快上市注册程序情况
创新是推动药品高质量发展的力量源泉,《药品注册管理办法》结合我国医药产业发展和临床需求实际,参考国际经验,设立了特别审批、突破性治疗药物、附条件批准、优先审评审批四个药品加快上市程序。《国家药监局关于发布<突破性治疗药物审评工作程序(试行)>等三个文件的公告》(2020年第82号),明确了加快通道的适用范围、适用条件、工作程序和政策支持等,既能显著提高相关程序执行过程中的可操作性,鼓励药物研制和创新,又能在全球抗击疫情的大背景下,依法依规对疫情防控所需药物实行特别审批,对于加快临床急需、临床价值突出、公共卫生急需等药物的上市具有重要推动作用。2020年已批准上市药品纳入加快上市程序情况详见附件4。
1.特别审批药物情况
在发生突发公共卫生事件的威胁时以及突发公共卫生事件发生后,国家药监局可依法决定对突发公共卫生事件应急所需防治药品实行特别审批。纳入实施特别审批程序的药物,国家药监局按照统一指挥、早期介入、快速高效、科学审批的原则,组织加快并同步开展药品注册受理、审评、核查、检验工作,并根据疾病防控的特定需要,限定其在一定的期限和范围内使用。
2020年新冠肺炎疫情在全球范围内不断蔓延,人民群众的生命安全受到严重威胁,药审中心闻令而动,第一时间科学、高效推进特别审评工作,按程序将59件中药、化学药、生物制品注册申请纳入特别审批程序并完成技术审评。建议附条件批准上市1件,为新型冠状病毒灭活**(Vero细胞);建议批准临床试验申请53件,其中5件已进入Ⅲ期临床试验,批准化湿败毒颗粒、清肺排毒颗粒的临床试验申请;批准了连花清瘟胶囊/颗粒、金花清感颗粒及血必净注射液等5件增加适应症的补充申请,加速了新冠病毒**和新冠肺炎治疗药物的上市进程,初步满足了新冠肺炎疫情防控的需要。
2.突破性治疗药物情况
突破性治疗药物指的是用于防治严重危及生命或者严重影响生存质量的疾病,且尚无有效防治手段或者与现有治疗手段相比有足够证据表明具有明显临床优势的创新药或者改良型新药等,申请人可在Ⅰ、Ⅱ临床试验阶段申请适用突破性治疗药物程序。根据《突破性治疗药物审评工作程序(试行)》,纳入到“突破性治疗”审评通道的药物,药审中心一是会优先处理相关沟通交流,加强指导并促进药物研发进程;二是在申报上市环节,该药物可适用优先审评审批程序,审评时限进一步缩短;三是上市申请阶段,药审中心会滚动接收其申报资料,并优先安排核查、检验等,可大大缩减新药从研发到上市的时间。2020年药审中心收到147件突破性治疗药物申请。经综合评估、公示,已将24件突破性治疗药物申请(21个品种)纳入突破性治疗药物程序,详见附件5。
3.附条件批准药物情况
附条件批准上市,目的在于缩短药物临床试验的研发时间,使其尽早应用于无法继续等待的危重疾病或公共卫生方面急需的患者。药物有效性评价的指标为临床终点,符合附条件批准上市情形的药物,可使用替代终点、中间临床终点或早期临床试验数据来反映药物的有效性,当这些数据能够提示药品的获益大于风险时候,即可申请附条件批准上市。
对于若不尽早进行治疗则会在数月或者更短时间内导致死亡的疾病患者来说,附条件批准上市的药物,使得这些无法继续等待的患者能够延续生命、提高生存质量,消除重大突发公共卫生事件对于人民生命安全的威胁。2020年药审中心审评通过的新药上市申请中,共有15件注册申请经附条件批准后上市,覆盖了新型冠状病毒感染引起的疾病、非小细胞肺癌、卵巢癌等适应症。
4.优先审评药物情况
(1)优先审评品种纳入情况
《药品注册管理办法》对优先审评审批程序的调整,是在多年实践经验基础上的优化,一是适用范围更多地向具有明显临床价值、临床急需和临床优势的药物聚焦,致力于将更多的临床价值显著、临床急需的短缺药品、防治重大传染病、罕见病、儿童用药、纳入突破性治疗程序、符合附条件批准的药品等纳入优先审评程序;二是审评时限的加速,药品上市许可申请的审评时限一般为200个工作日,与完整的申报路径相比,优先审评审批程序的审评时限缩短至130个工作日,其中临床急需境外已上市罕见病用药优先审评审批程序的审评时限为70个工作日。药审中心通过优化审评资源配置率,在高标准完成技术审评的前提下,力争按时限完成审评,推动纳入优先审评审批程序中的品种尽快获批上市。
根据《药品注册管理办法》、46号公告、《国家食品药品监督管理总局关于鼓励药品创新实行优先审评审批的意见》(食药监药化管〔2017〕126号,以下简称126号文件),2020年药审中心将219件(按通用名计127个品种)注册申请纳入优先审评审批程序。其中,144件注册申请按照126号文件规定的范围纳入优先审评审批程序,75件按照《药品注册管理办法》规定的范围纳入优先审评审批程序,包括42件儿童用药和罕见病用药。2016-2020年纳入优先审评审批程序的各类注册申请情况详见表6和表7。
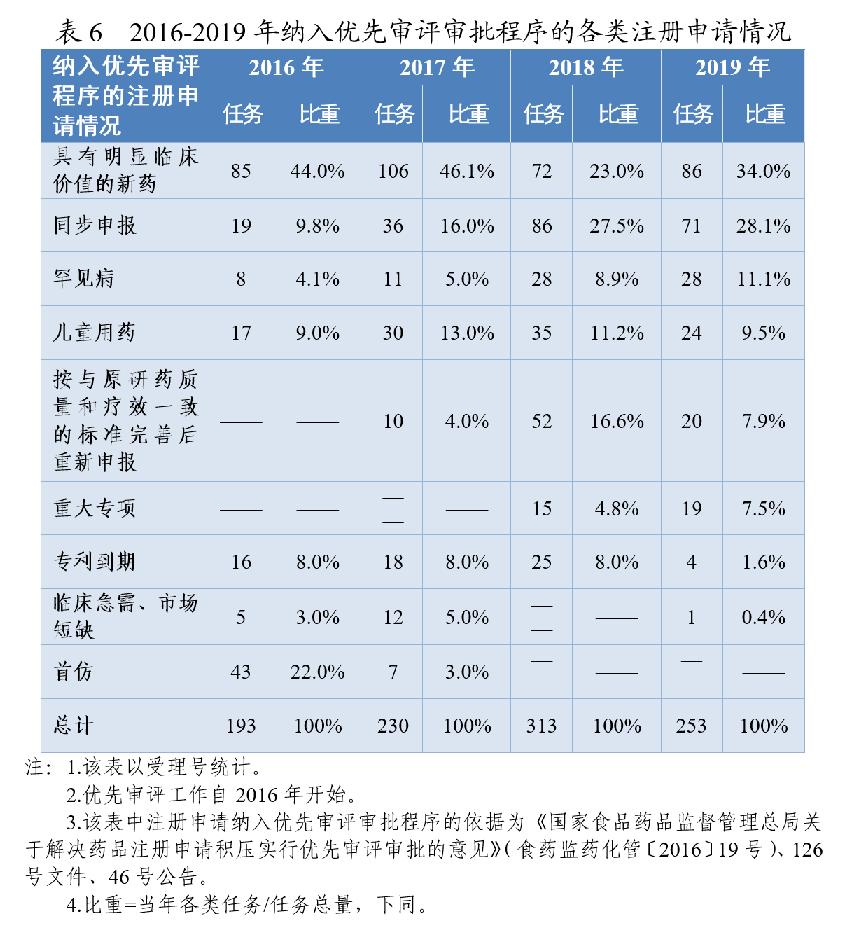
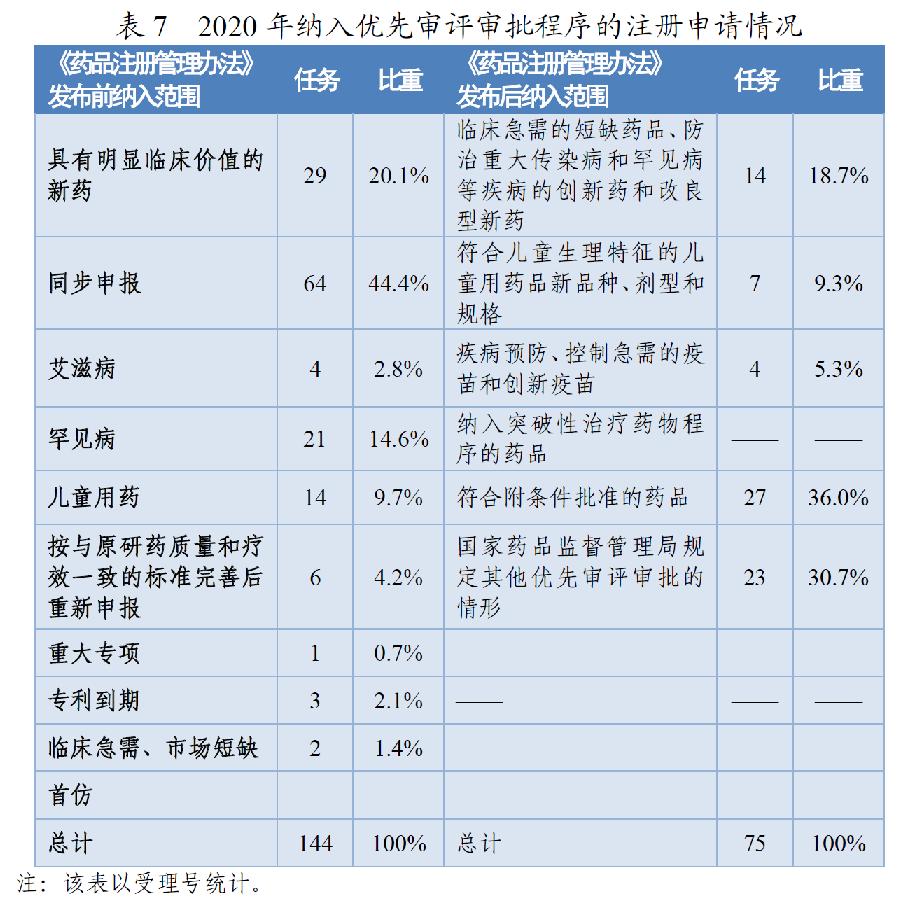
按此前优先审评范围纳入的注册申请中,同步申报占比多达44%(64/144),具有明显临床价值的新药占比为20%,按与原研药质量和疗效一致的标准完善后重新申报品种占比则由7.9%降至4.2%。
按照《药品注册管理办法》优先审评范围纳入的注册申请中,符合附条件批准的药品占比为36%(27/75),创新药和儿童用药占比28%(21/75),优先审评资源已向具有明显临床价值的创新、急需药物倾斜。
(2)优先审评品种完成情况
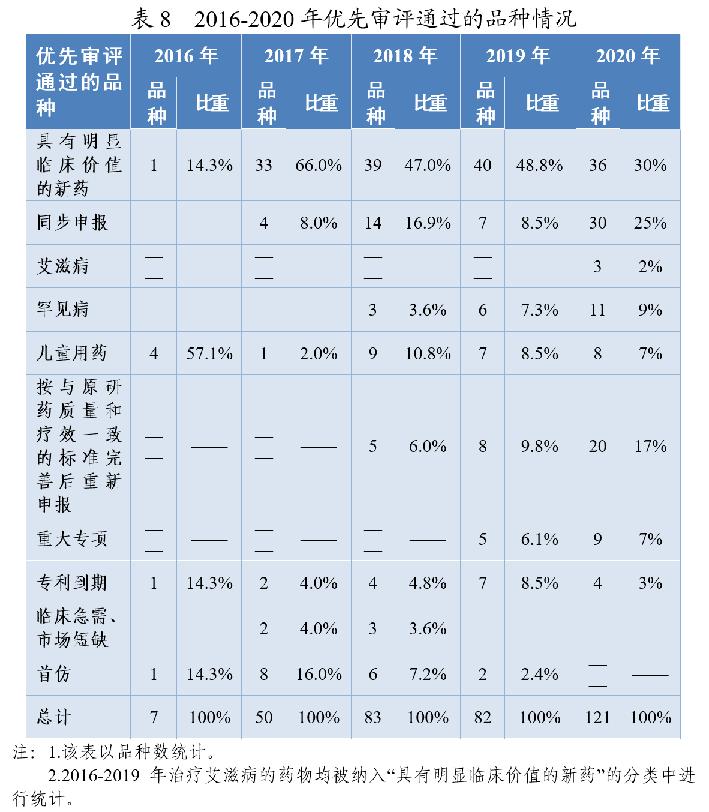
2020年有217件注册申请(按通用名计121个品种)通过优先审评程序建议批准上市(含已上市药品新增适应症),审评通过件数较2019年增长51.7%,例如:我国自主研发的1类创新药甲磺酸阿美替尼片、泽布替尼胶囊、奥布替尼片等,治疗罕见病法布雷病阿加糖酶α注射用浓溶液,用于配合饮食控制及运动治疗2型糖尿病的中药新药桑枝总生物碱片、间变性淋巴瘤激酶抑制剂盐酸恩沙替尼胶囊、成人复发型多发性硬化治疗药物西尼莫德片等。2016-2020年优先审评通过的品种情况详见表8。
(八)药品注册现场核查相关情况
1.总体情况
药审中心积极落实《药品注册管理办法》,转变药品注册核查理念,将注册现场核查启动工作模式由基于审评需要调整为基于风险启动,并联开展技术审评与注册现场核查工作;对于自2020年7月1日起受理的注册申请,在受理后40个工作日内决定是否启动相应注册现场核查任务。为便于申请人及时获知注册现场核查启动相关信息,完善药审中心网站申请人之窗栏目,开通递交注册现场核查用生产工艺与质量标准通道和查收注册现场核查电子通知函的功能。
2.注册现场核查具体情况
2020年,药审中心基于品种因素和研发生产主体合规因素风险启动注册现场核查任务1235个。其中,药学现场核查任务792个,临床试验数据核查任务439个,药理毒理研究核查任务4个。
药审中心接收核查报告818份。其中,药学现场核查报告449份,临床试验核查报告363份,药理毒理研究核查报告6份。
(九)沟通交流情况
1.总体情况
2020年,药审中心在落实疫情防控要求的同时,尽量满足申请人的需要,全力保障各类沟通交流畅通。在推动新冠病毒**和新冠肺炎治疗药物的研发方面,为79个新冠病毒**,中医药、中和抗体(27个)等新冠肺炎治疗药物,组织申请人与药审中心审评团队之间的沟通交流5600余次,并针对新冠病毒**、中和抗体等重点品种,单独设立了台账,动态跟进;在维护与申请人沟通桥梁方面,药审中心发布了《药物研发与技术审评沟通交流管理办法》《药审中心关于业务咨询服务联络方式的通知》,优化了电话咨询服务,每天有专人接听解答申请人咨询电话,根据咨询问题类型的不同设立了8个联系邮箱,及时解答处理申请人问题,不断提高沟通交流的质量和效率。
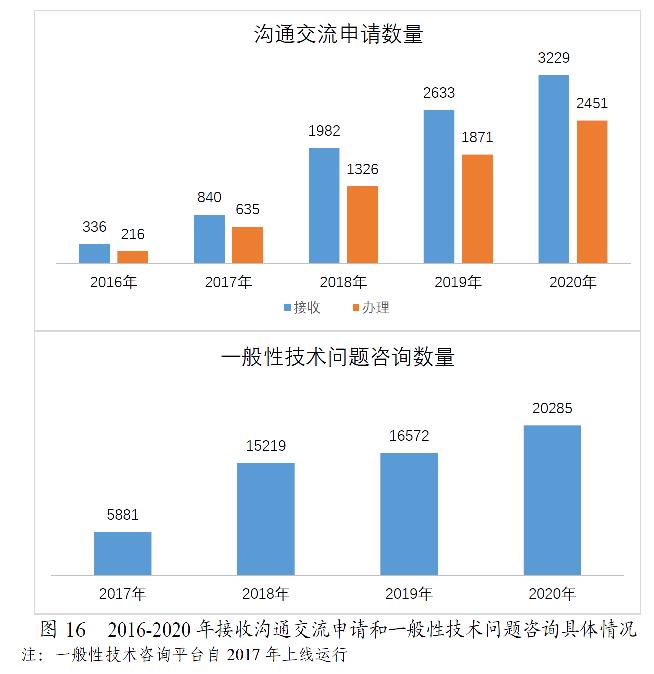
药审中心接收沟通交流会议申请3229件,较2019年增长22.64%,办理沟通交流会议申请2451件,较2019年增长31.00%。在网络平台接收一般性技术问题咨询20285个,较2019年增长22.41%;接收电话咨询超过上万次,邮件咨询数千件,同时也面向社会提供现场咨询服务。2016-2020年接收沟通交流申请和一般性技术问题咨询具体情况详见图16。
2.沟通交流会议申请的完成情况
药审中心所接收的3229件沟通交流会议申请中,符合会议召开条件的,及时与申请人取得了联系,商议会议细节;无需召开会议的,以书面形式尽快回复了申请人。2020年共办理了2451件沟通交流会议申请。在药物研发关键阶段召开的Ⅱ类会议占比76.42%,其中临床前(Pre-IND)申请占比37.49%。2020年各类沟通交流会议申请及办理情况详见表9。
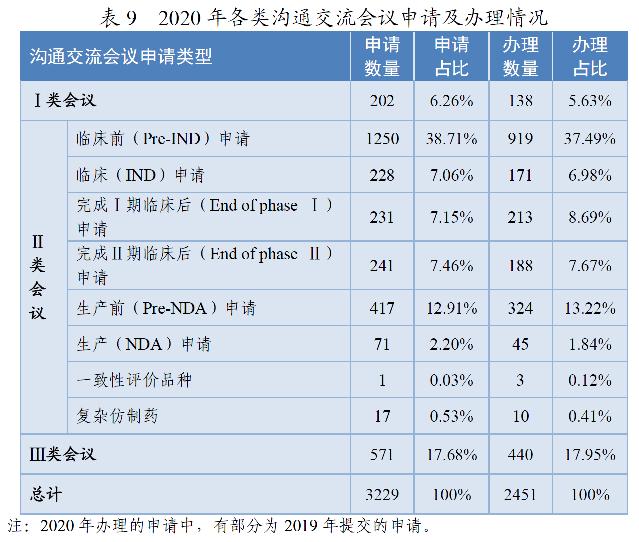
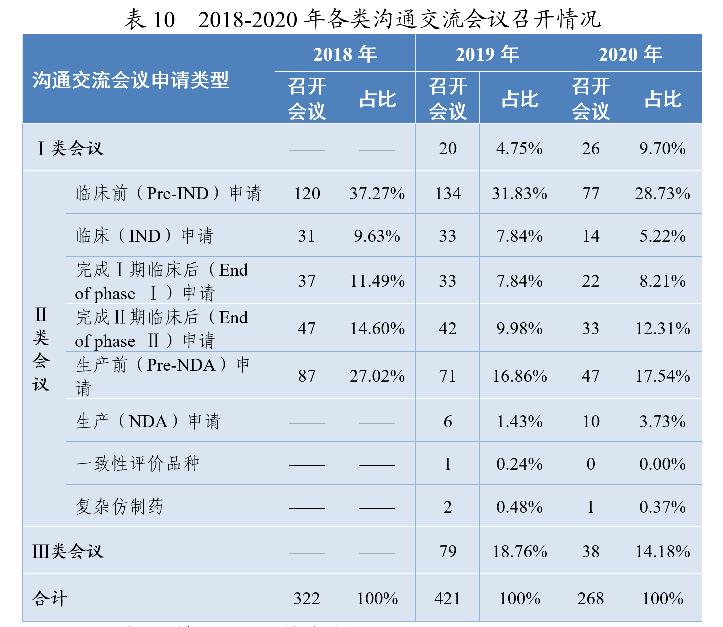
沟通交流会议的形式为电话会议、视频会议、面对面会议,共召开沟通交流会议268次,以书面形式回复两千余件。2018-2020年各类沟通交流会议的召开情况详见表10。
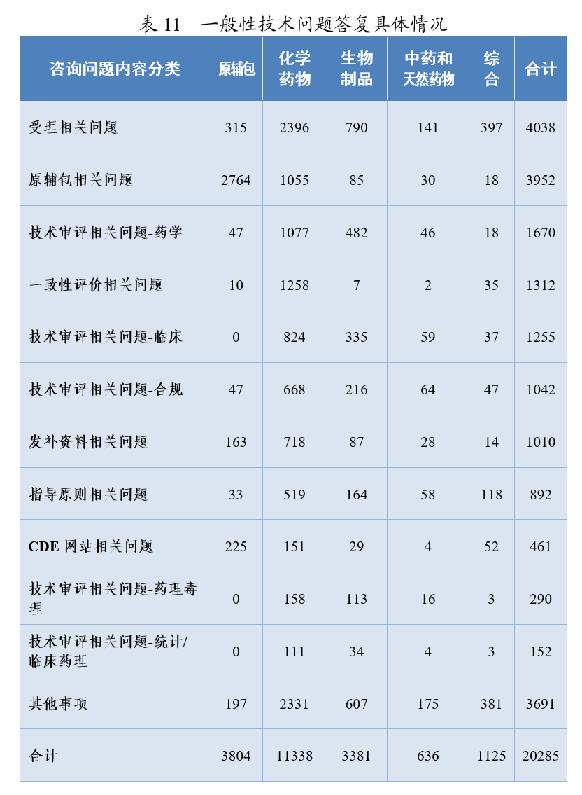
3.一般性技术问题答复情况
药审中心通过网上咨询平台共接收了20285个一般性技术问题的咨询。按照内容分类,问题主要集中于受理(4038个)、原辅包(3952个)等方面;按照药品分类,问题主要集中于化学药(11338个)方面,其中化学药受理(2396个)、化学药一致性评价(1258个)。一般性技术问题答复具体情况详见表11。
二、药品注册申请受理情况
(一)总体情况
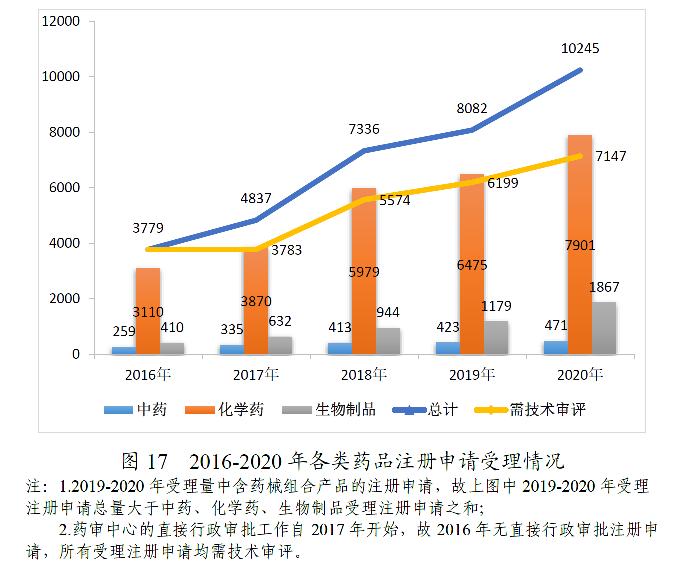
2020年,根据46号公告、《国家药监局关于发布生物制品注册分类及申报资料要求的通告》(2020年第43号)、《国家药监局关于发布化学药品注册分类及申报资料要求的通告》(2020年第44号)、《国家药监局关于发布<中药注册分类及申报资料要求>的通告》(2020年第68号)等,药审中心受理中药、化学药、生物制品各类注册申请共10245件(含药械组合产品6件),较2019年增长26.76%。其中,需技术审评的注册申请7147件(含5695件需药审中心技术审评和行政审批的注册申请),较2019年增长15.29%;直接行政审批的注册申请3092件,较2019年增长64.64%。
受理的10239件药品注册申请中,化学药注册申请受理量为7901件,较2019年增长22.02%,占2020年全部注册申请受理量的77.17%,2016-2020年各类药品注册申请受理情况详见图17。
2.药审中心的直接行政审批工作自2017年开始,故2016年无直接行政审批注册申请,所有受理注册申请均需技术审评。
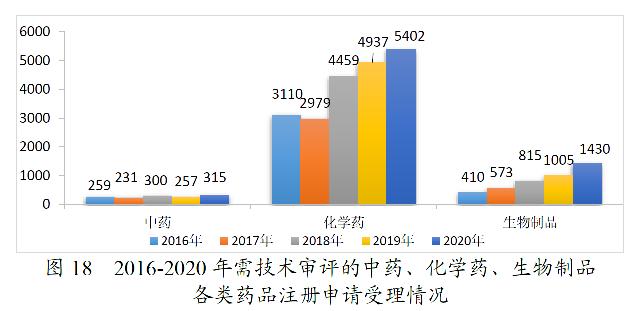
2020年受理的需技术审评的注册申请7147件中,化学药注册申请为5402件,较2019年增长9.42%,占全部需技术审评的注册申请受理量的75.58%;中药注册申请315件,较2019年增长22.57%;生物制品注册申请1430件,较2019年增长42.29%。2016-2020年需技术审评的中药、化学药、生物制品各类注册申请受理情况详见图18。
(二)1类创新药受理情况
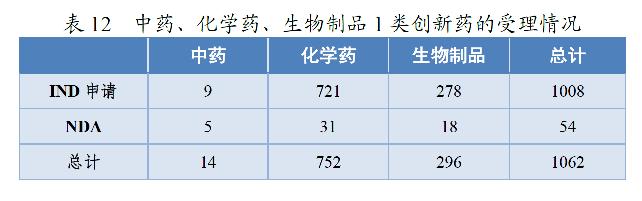
2020年,药审中心受理1类创新药注册申请共1062件(597个品种),较2019年增长51.71%。其中,受理IND申请1008件(559个品种),较2019年增长49.78%;受理NDA 54件(38个品种),较2019年增长100.00%。以药品类别统计,中药、化学药、生物制品1类创新药受理量分别为14、752、296件。以生产场地统计,境内生产药品843件,境外生产药品219件。详见表12和13。
(三)各类注册申请受理情况
1.中药注册申请受理情况
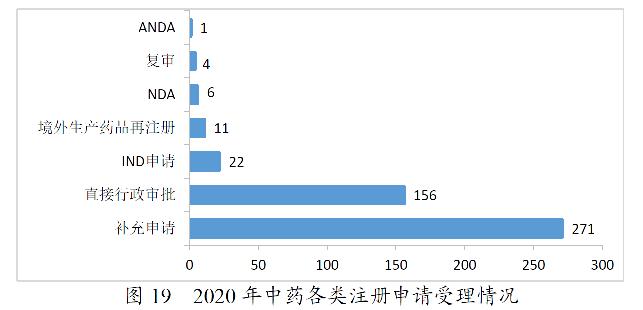
2020年,药审中心受理中药注册申请471件。其中,受理中药IND申请22件,受理中药NDA 6件,受理中药ANDA 1件。2020年中药各类注册申请受理情况详见图19。
受理1类中药创新药注册申请14件。其中,受理IND 申请9件(9个品种),受理NDA 5件(5个品种)。
2.化学药注册申请受理情况
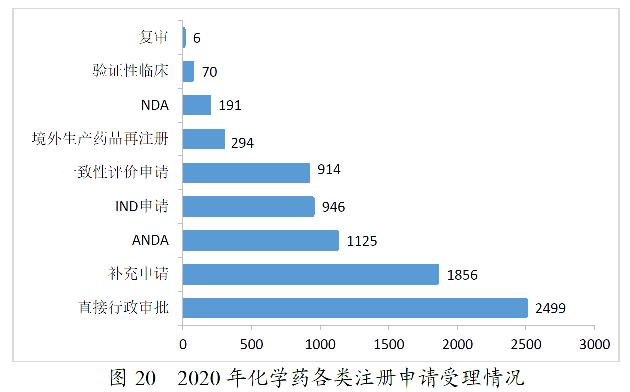
2020年,药审中心受理化学药注册申请7901。其中,受理化学药IND申请946件,较2019年增长36.31%;受理化学药NDA 191件,较2019年增长46.92%;受理ANDA 1125件,较2019年增长7.45%;受理一致性评价申请914件,较2019年减少11.95%。2020年化学药各类注册申请受理情况详见图20。2016-2020年化学药各类注册申请受理情况详见图21。
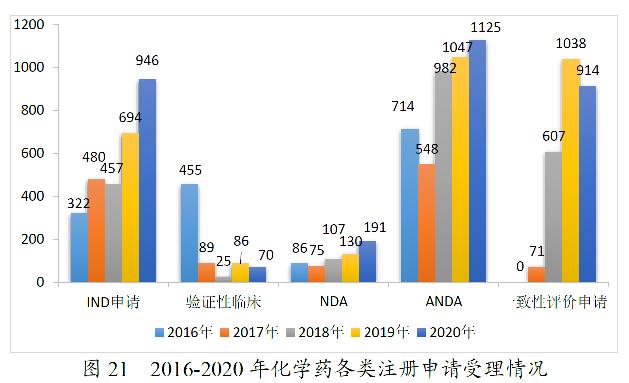
药审中心受理1类创新化学药注册申请752件(360个品种),较2019年增长31.24%。其中,受理IND申请721件(339个品种),较2019年增长30.62%;受理NDA 31件(21个品种),较2019年增长47.62%。
药审中心受理化学药5.1类注册申请160件,较2019年增长1.91%。其中受理临床试验申请(验证性临床)53件,受理NDA 107件。
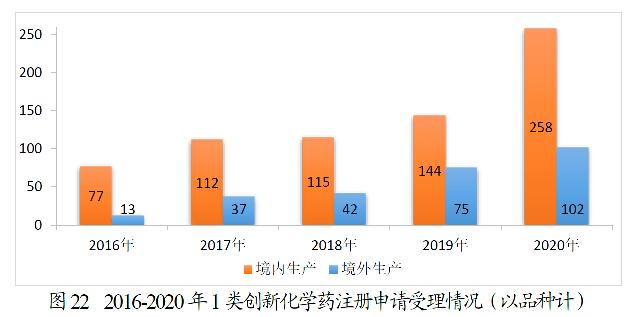
360个品种的1类创新化学药注册申请中,境内生产化学药注册申请为258个品种,境外生产化学药注册申请为102个品种。2016-2020年1类创新化学药注册申请受理情况详见图22。
3.生物制品注册申请受理情况
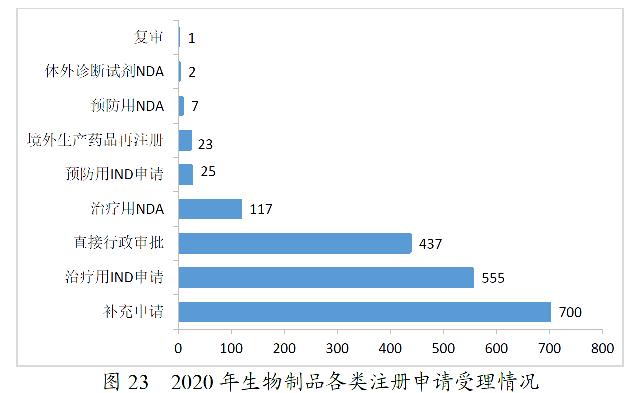
2020年,药审中心受理生物制品注册申请1867件。其中,受理生物制品IND申请580件(预防用IND申请25件,治疗用IND申请555件),较2019年增长87.10%;受理生物制品NDA 126件(预防用NDA 7件,治疗用NDA 117件,体外诊断试剂2件),较2019年增长1.62%。2020年生物制品各类注册申请受理情况详见图23。2016-2020年生物制品IND申请和NDA受理情况详见图24。
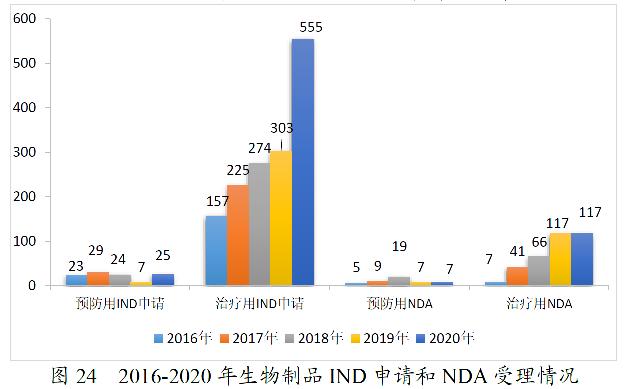
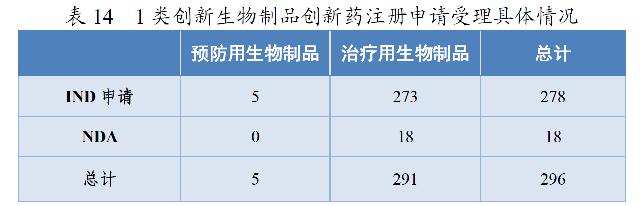
药审中心受理1类创新生物制品注册申请296件(223个品种),较2019年增长133.07%。其中,受理预防用生物制品5件,受理治疗用生物制品291件;受理生物制品IND申请278件(211个品种),较2019年增长129.75%;受理生物制品NDA 18件(12个品种),较2019年增长200.00%,具体情况详见表14。
4.行政审批注册申请受理情况
(1)总体情况
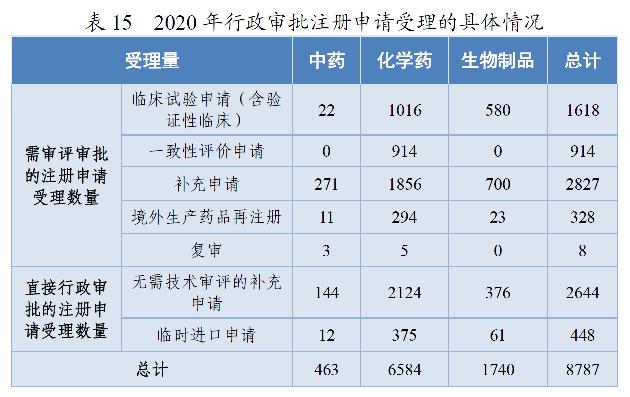
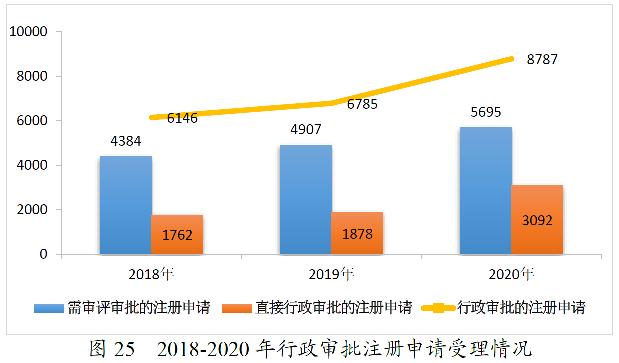
2020年,药审中心受理需中心行政审批的中药、化学药、生物制品各类注册申请8787件,较2019年增长29.51%。其中,受理需审评审批的注册申请(临床试验申请、一致性评价申请、补充申请、境外生产药品再注册及复审)5695件,较2019年增长16.06%;受理直接行政审批的注册申请(无需技术审评的补充申请、临时进口申请)3092件,较2019年增长64.64%。2020年行政审批注册申请受理的具体情况详见表15。2016-2020年行政审批注册申请受理情况详见图25。
(2)需审评审批的注册申请受理情况
药审中心受理5695件需审评审批的注册申请中,临床试验申请1618件(含验证性临床)、一致性评价申请914件、补充申请2827件、境外生产药品再注册328件、复审8件。
(3)直接行政审批的注册申请受理情况
药审中心受理3092件直接行政审批的注册申请中,按申请类型划分,补充申请2644件、临时进口申请448件。按药品类型划分,中药156件、化学药2499件、生物制品437件。
三、重点治疗领域品种
新冠病毒**和新冠肺炎治疗药物:
1.新型冠状病毒灭活**(Vero细胞),为国内首 个附条件批准的新冠病毒**,也是全球首 个新冠病毒灭活**。适用于18岁及以上人群预防由新型冠状病毒(SARS-CoV-2)感染引起的疾病。
2.“三药”品种,为《新型冠状病毒肺炎诊疗方案(试行)》推荐药物,即连花清瘟颗粒/胶囊、金花清感颗粒和血必净注射液。连花清瘟颗粒/胶囊和金花清感颗粒新增适应症用于在新型冠状病毒肺炎的常规治疗中的轻型、普通型引起的发热、咳嗽、乏力,血必净注射液新增适应症用于新型冠状病毒肺炎重型、危重型的全身炎症反应综合征或/和多脏器功能衰竭,其获批上市充分发挥了中医药在疫情防控中的作用。
3.注射用西维来司他钠,为中性粒细胞弹性蛋白酶选择性抑制剂,适用于改善伴有全身性炎症反应综合征的急性肺损伤/急性呼吸窘迫综合征(ALI/ARDS),是全球唯一用于ALI/ARDS的药物,其获批上市填补了我国ALI/ARDS药物治疗领域的空白,为我国呼吸系统危重症患者提供用药选择。
抗肿瘤药物:
4.甲磺酸阿美替尼片,为我国首 个具有自主知识产权的第三代靶向表皮生长因子受体(EGFR)小分子酪氨酸激酶抑制剂(TKI)创新药物,适用于治疗既往经EGFR-TKI治疗时或治疗后出现疾病进展,并且经检测确认存在EGFR T790M突变阳性的局部晚期或转移性非小细胞肺癌。本品疗效突出,脑转移病灶控制良好,其获批上市将显著改善该疾病治疗药物的可及性。
5.索凡替尼胶囊,为多靶点、抗血管生成口服小分子酪氨酸激酶抑制剂,是国内首 个用于治疗无法手术切除的局部晚期或转移性、进展期非功能性、分化良好(G1、G2)的非胰 腺来源的神经内分泌瘤的创新药物。本品疗效突出,显著降低了此类患者的疾病进展和死亡风险,其获批上市填补了该疾病治疗领域的空白。
6.注射用维布妥昔单抗,为全球首 个CD30靶点抗体偶联药物(ADC),也是国内首 个用于恶性淋巴瘤患者的ADC药物,适用于治疗复发或难治性的系统性间变性大细胞淋巴瘤和经典型霍奇金淋巴瘤,本品获批上市为改善我国此类患者的长期生存提供了有效的治疗手段。
7.注射用贝林妥欧单抗,为全球首 个双特异性抗体(CD3和CD19靶点)药物,也是我国首 个用于肿瘤适应症的双特异性抗体药物,适用于治疗成人复发或难治性前体B细胞急性淋巴细胞白血病。对于化疗失败的复发或难治性急性淋巴细胞白血病患者,与标准化疗相比,本品可显著延长患者生存期,其获批上市为我国此类患者提供了更好的治疗手段。
8.甲磺酸仑伐替尼胶囊,为多靶点、口服酪氨酸激酶抑制剂,是国内首 个用于治疗进展性、局部晚期或转移性**碘难治性分化型甲状腺癌的小分子药物。本品疗效突出,其获批上市为我国此类患者提供了有效的治疗方案,填补了该治疗领域的空白。
抗感染药物:
9.盐酸可洛派韦胶囊,为非结构蛋白5A(NS5A)抑制剂,是我国具有自主知识产权的广谱、直接抗丙肝病毒创新药物,适用于与索磷布韦联用治疗初治或干扰素经治的基因1、2、3、6型成人慢性丙型肝炎病毒感染,可合并或不合并代偿性肝硬化,本品获批上市为我国慢性丙肝患者提供了一种新的治疗选择。
10.恩曲他滨替诺福韦片,增加适应症用于降低成人和青少年(体重至少在35 kg以上)通过高风险性行为获得HIV-1的风险,是国内首 个用于暴露前预防HIV的药物。HIV感染是重大公共卫生问题,本品获批上市对于控制HIV传播具有重大意义。
循环系统药物:
11.拉那利尤单抗注射液,为全人源化单克隆抗体(IgG1/K-轻链),是我国首 个用于12岁及以上患者预防遗传性血管性水肿(HAE)发作的药物。HAE疾病反复发作,近半数患者可出现上呼吸道黏膜水肿引发窒息而危及生命,本品获批上市为我国HAE患者预防发作提供了安全有效的治疗手段。
12.氯苯唑酸软胶囊,为转甲状腺素蛋白(TTR)稳定剂,适用于治疗转甲状腺素蛋白淀粉样变性心肌病,以减少心血管死亡及心血管相关住院。该疾病是一种致命性疾病,属罕见病,本品为我国首 个针对该病病因治疗的药物,其获批上市为我国此类患者提供了新的治疗手段。
呼吸系统药物:
13.苯环喹溴铵鼻喷雾剂,为胆碱能受体拮抗剂,为我国首 个具有自主知识产权用于变应性鼻炎的鼻用抗胆碱创新药物,适用于改善变应性鼻炎引起的流涕、鼻塞、鼻痒和喷嚏症状,本品其获批上市可为我国此类患者提供新的治疗选择。
14.乙磺酸尼达尼布软胶囊,为小分子酪氨酸激酶抑制剂,具有抗纤维化作用,增加适应症用于治疗系统性硬化病相关间质性肺疾病(SSc-ILD)和具有进行性表型的慢性纤维化性间质性肺疾病(PF-ILD)。目前可用于SSc-ILD和PF-ILD的有效治疗方式有限,临床用药需求迫切,本品获批新增适应症可以填补该治疗领域空白,为我国此类患者提供药物选择。
神经系统药物:
15.氘丁苯那嗪片,为囊泡单胺转运蛋白2(VMAT2)抑制剂,是我国首 个用于治疗与罕见病亨廷顿病有关的舞蹈病、迟发性运动障碍的药物,属临床急需境外新药名单品种,本品获批上市满足了我国此类患者迫切的临床需求。
16.氯苯唑酸葡胺软胶囊,为转甲状腺素蛋白(TTR)稳定剂,是我国首 个用于治疗成人转甲状腺素蛋白淀粉样变性多发性神经病I期症状患者、延缓周围神经功能损害的药物,属临床急需境外新药名单品种,其获批上市改变了该病无药可治的局面。
镇痛药及**科药物:
17.环泊酚注射液,为GABAA受体激动剂,是用于消化道内镜检查中镇静的创新药物。本品与临床常用**镇静药物丙泊酚具有相似的药理机制,但具有起效快,注射痛少,呼吸抑制轻,恢复速度快等优势特征,其获批上市可为我国消化内镜检查操作用药提供新的选择。
皮肤五官药物:
18.塞奈吉明滴眼液,为国内首 个用于治疗神经营养性角膜炎(NK)的重组人神经生长因子(rhNGF)药物,属临床急需境外新药名单品种。NK为罕见的退行性角膜疾病,可致盲,中重度NK手术治疗为侵入性操作,费用高且不能永 久治愈,本品获批上市为我国此类患者提供了有效的治疗药物,预计将成为中重度NK患者的首选治疗。
19.度普利尤单抗注射液,为重组人免疫球蛋白-G4单克隆抗体,适用于治疗外用处方药控制不佳或不建议使用外用处方药的成人中重度特应性皮炎,属临床急需境外新药名单品种。与现有治疗方式相比,本品有明显临床优势,其获批上市为此类难治性严重疾病患者提供了治疗选择。
消化系统药物:
20.注射用维得利珠单抗,为作用于人淋巴细胞整合素α4β7的人源化单克隆抗体,适用于治疗对传统治疗或肿瘤坏死因子α(TNF-α)抑制剂应答不充分、失应答或不耐受的中度至重度活动性溃疡性结肠炎、克罗恩病,属临床急需境外新药名单品种。此类疾病存在迫切的临床治疗需求,特别是对于TNF-α拮抗剂治疗失败的患者,本品获批上市可为临床提供新的治疗选择。
外科药物:
21.注射用丹曲林钠,适用于预防及治疗恶性高热(MH),是目前唯一短时间内给药可改变该疾病转归的药物。MH临床结局危重,死亡率高,其获批上市可改变目前国内MH无安全有效治疗手段的现状,满足迫切临床需求。
22.他克莫司颗粒,适用于预防儿童肝 脏或肾 脏移植术后的移植物排斥反应,治疗儿童肝 脏或肾 脏移植术后应用其他免疫抑制药物无法控制的移植物排斥反应,属儿童用药,本品获批上市可极大解决我国儿科肝肾移植患者未满足的临床需求。
罕见病药物:
23.注射用拉罗尼酶浓溶液,为国内首 个用于罕见病黏多糖贮积症I型(MPS I,α-L-艾杜糖苷酶缺乏症)的酶替代治疗药物,属临床急需境外新药名单品种。黏多糖贮积症I型是一种严重危及生命且国内尚无有效治疗手段的遗传性罕见病,已列入我国第一批罕见病目录,本品获批上市填补了我国此类患者的用药空白。
24.艾度硫酸酯酶β注射液,为国内首 个用于罕见病黏多糖贮积症Ⅱ型(MPS Ⅱ,亨特综合征)的酶替代治疗药物。黏多糖贮积症Ⅱ型是一种严重危及生命且国内尚无有效治疗手段的遗传性罕见病,已列入我国第一批罕见病目录,本品获批上市填补了我国此类患者的用药空白。
体内诊断试剂:
25.重组结核杆菌融合蛋白(EC),适用于6月龄及以上婴儿、儿童及65周岁以下成人结核杆菌感染诊断,并可用于辅助结核病的临床诊断,为全球首 个用于鉴别卡介苗接种与结核杆菌感染的体内诊断产品,其获批上市为临床鉴别诊断提供了新的手段。
预防用生物制品(**):
26.鼻喷冻干流感减毒活**:为国内首家以鼻喷途径接种的**,适用于3(36月龄)~17岁人群用于预防由**相关型别的流感病毒引起的流行性感冒,接种后可刺激机体产生抗流感病毒的免疫力。
中药新药:
27.桑枝总生物碱片,其主要成分为桑枝中提取得到的桑枝总生物碱,是近10年来首 个获批上市的抗糖尿病中药新药,适用于配合饮食控制及运动、治疗2型糖尿病。本品可有效降低2型糖尿病受试者糖化血红蛋白水平,其获批上市为2型糖尿病患者提供新的治疗选择。
28.筋骨止痛凝胶,为醋延胡索、川芎等12种药味组成的中药复方新药,适用于膝骨关节炎肾虚筋脉瘀滞证的症状改善,具有“活血理气,祛风除湿,通络止痛”的功效。本品为外用凝胶制剂,药物中各成分通过透皮吸收而发挥作用,可避免肠胃吸收和肝 脏首过代谢,其获批上市可为膝关节骨性关节炎患者提供新的治疗选择。
29.连花清咳片,为麻黄、桑白皮等15种药味组成的中药新药,适用于治疗急性气管-支气管炎痰热壅肺证引起的咳嗽、咳痰等,具有“宣肺泄热,化痰止咳”的功效,其获批上市可为急性气管-支气管炎患者提供新的治疗选择。
四、全力做好应急审评工作
(一)加强统一领导,制定工作程序
按照国家药监局党组关于疫情防控应急审评审批工作部署,药审中心闻令而动,一是充分发挥集体决策作用,迅速成立抗新型冠状病毒 药物特别审评领导小组,抽调16个部门148名骨干力量为工作小组成员,先后召开特别审评领导小组会议6次和领导小组专题会18次,明晰工作原则,优化工作流程,及时研究解决应急审评过程中遇到的问题,保证工作顺利推进、有序开展。二是制定工作程序,形成1个方案、2个程序、1个规范,即《药审中心抗新型冠状病毒 药物特别审评工作方案》《关于新型冠状病毒(2019-nCoV)药物立项申请评议工作程序》《关于新型冠状病毒(2019-nCoV)药物特别专家组评估和审核工作程序》《国家药监局抗新型冠状病毒 药物专家会议管理规范》。三是严格落实“安全守底线、疗效有证据、质量能保证、审评超常规”的工作要求,按照工作程序,依法依规、科学规范审评,标准不降,加速审批。
(二)发挥专家作用,解决技术难题
一是组建特别专家组。按照《国家食品药品监督管理局药品特别审批程序》(原国家食品药品监督管理局令第21号)规定和国家药监局新型冠状病毒感染肺炎疫情应对工作组药品组的决定,药审中心先后遴选出多位院士和知名专家组成了特别专家组,经国家药监局批准后开展工作。遇到新的技术难点问题时,听取专家意见建议后,由专家投票表决。二是注重发挥专家技术支撑作用。通过专家研讨会和专家咨询会解决特定技术问题,例如针对mRNA新冠病毒**在研发上存在的难点和潜在的风险,药审中心组织专家形成技术指导原则,以指导相关企业的研发。
(三)实时高效沟通,提高研发进度
一是遵循“早期介入、持续跟进、主动服务”的工作要求,第一时间组织审评力量对咨询品种或注册申请立项的科学性和可行性进行评议,并在24小时内与申请人进行沟通交流,保证申请人尽快提交特别审批注册申请。二是加强国际合作。积极参加世界卫生组织(WHO)、国际药品监管机构联盟(ICMRA)等组织召开的视频电话会议,共同探讨研发审评标准,了解新冠病毒**研发信息,指导推动研发企业赴国外开展Ⅲ期临床试验。
(四)探索研审联动,坚持科学审评
一是探索建立研发审评联动工作机制。边研发、边提交、边审评,为新冠病毒**研发争取到了宝贵时间,确保新冠病毒**等研发走在世界前列。通过这种工作机制,大大缩短了审评时间。二是建立技术标准体系。针对新冠病毒的特点,及时制定新冠病毒**、新冠肺炎治疗药物研发技术指导原则等28个技术文件,指导企业高标准研发,少走弯路,科学开展审评。
五、鼓励中药传承创新发展
贯彻落实习近平总书记关于中医药的重要指示**、《中共中央 国务院关于促进中医药传承创新发展的意见》及国家药监局要求,药审中心从改革中药注册分类、健全中药技术指导原则等各方面积极鼓励中药守正创新。一是推动中药的传承发展。起草并由国家药监局发布《中药注册分类及申报资料要求》,丰富古代经典名方复方制剂的范围,促进古代经典名方中药复方制剂研发,推动其向新药转化。二是建立完善符合中药特点的质量控制体系。遵循中药特点和研发规律,将中药独特的理论体系和实践特点、中药制剂质量控制特点与药品质量控制的一般要求有机结合,研究构建完善符合中药制剂特点的质量控制方法和策略,制定《中药新药用饮片炮制研究指导原则(试行)》《中药新药质量标准研究技术指导原则(试行)》《中药复方制剂生产工艺研究技术指导原则(试行)》《中药生物效应检测研究技术指导原则(试行)》等8个技术指导原则。三是健全符合中药特点的审评体系。引入新工具、新方法、新标准用于中药疗效评价,细化申报资料要求,制定《中药新药用于慢性便秘临床研究技术指导原则》《中药新药用于糖尿病肾病临床研究技术指导原则》等技术指导原则,探索构建中医药理论、人用经验和临床试验相结合的审评证据体系。四是全力做好中药特别审评工作。充分发扬抗疫**,制定了《用于新冠肺炎中药注册申请特别审批申报资料要求(试行)》《用于新冠肺炎中药注册申请特别审批技术指导原则(试行)》等,指导应急状态下的中药审评。截至2020年12月31日,“三方”中的清肺排毒颗粒、化湿败毒颗粒的IND申请已获批准,“三药”连花清瘟颗粒/胶囊、金花清感颗粒、血必净注射液获批增加用于治疗新冠肺炎的适应症。五是赴武汉开展实地调研和座谈,持续推进中药监管科学“以临床价值为导向的中药安全性评价研究”课题研究。六是开展援疆援藏工作,赴西藏开展实地调研、与新疆维吾尔自治区药品监督管理局召开线上座谈交流会,支持民族药发展。
六、加强《药品注册管理办法》配套文件制修订
新修订的《药品注册管理办法》是贯彻党中央、国务院审评审批制度改革**和落实新修订《药品管理法》的重要规章,考虑到药品注册管理中的具体技术要求,需要跟随技术发展的脚步不断调整完善,在规章中不适宜作出具体的规定,因此这些具体技术要求在《药品注册管理办法》发布后,以配套文件、技术指导原则等形式发布,既能更好地顺应药品研发的科学规律,也能确保新旧《药品注册管理办法办法》的平稳过渡和新《药品注册管理办法》的顺利实施。
根据国家局部署,药审中心统筹协调,加大配套文件的制修订力度,成立课题组,对重点难点问题开展调研攻关,充分听取专家、业界意见,力求达成共识,共参与了48个配套文件制修订工作,其中牵头起草配套文件30个。自开展工作以来,已基本完成配套文件公开征求意见工作,部分文件已经国家局审核同意后发布实施,有效确保了各项审评任务不断、不散、不乱,新旧注册相关规定的顺利过渡和实施。
七、加快审评技术标准体系建设
在药品审评和研发过程中,指导原则兼具监管依据和技术要求的双重职能。《药品注册管理办法》明确从事药物研制和药品注册活动,应当遵守有关法律、法规、规章、标准和规范;药审中心等专业技术机构,应当根据科学进展、行业发展实际和药品监督管理工作需要制定技术指导原则和程序,并向社会公布。
药品技术指导原则体系的建立与完善,是落实“四个最严”要求的最好实践,是药审中心推进审评体系和审评能力现代化的重要举措。药审中心通过“定标准、定程序、定计划”三步走的方式,统筹规划以指导原则为核心的审评标准体系建设,围绕药品研发需求和鼓励创新的原则,对标国际先进监管机构技术标准,加大指导原则制定和公开力度。2020年共开展了119个技术指导原则制修订工作,根据《国家药监局综合司关于印发药品技术指导原则发布程序的通知》(药监综药管〔2020〕9号)要求,截至2020年12月31日,药审中心已经国家药监局审查同意发布了71个指导原则,详见附件6。
在应对新型冠状病毒肺炎、促进新冠病毒**和新冠肺炎治疗药物的研发和审评质量、速度方面,药审中心发布了《新型冠状病毒预防用**研发技术指导原则(试行)》等7个指导原则;在着力提升中药材质量研究,鼓励中药研发与创新方面,发布了《中药新药用药材质量控制研究技术指导原则(试行)》《中药复方制剂生产工艺研究技术指导原则(试行)》《中药新药用于慢性便秘临床研究技术指导原则》等10个指导原则;在鼓励儿童药物研发方面,发布了《真实世界研究支持儿童药物研发与审评的技术指导原则(试行)》等3个指导原则;在支持抗肿瘤药物研发,进一步满足申请人对具体抗肿瘤药物的个药指导原则需求方面,发布了《抗肿瘤药联合治疗临床试验技术指导原则》《注射用曲妥珠单抗生物类似药临床试验指导原则》等22个指导原则;在提高仿制药质量,推进仿制药一致性评价方面,规范审评标准和尺度,发布了《化学药品注射剂仿制药质量和疗效一致性评价技术要求》《化学药品注射剂(特殊注射剂)仿制药质量和疗效一致性评价技术要求》等9个指导原则。这些指导原则覆盖新冠应急审评标准、儿童用药、中药民族药技术标准体系、抗肿瘤药物研发及仿制药研发等热点难点问题,持续完善药品技术指导原则体系,有效推动药物研发创新,不断优化统一审评尺度,大力提升审评质量和效率,显著减少审评自由裁量权。
八、持续深化药品审评审批制度改革
(一)加快境外已上市临床急需新药审评
深入贯彻国务院常务会议**,落实加快境外已上市临床急需新药审评要求,在确定了第一二批74个品种名单的基础上,国家药监局、国家卫生健康委组织有关专家研究论证,遴选临床急需新药品种,药审中心发布了第三批7个品种名单。对于符合《国家药品监督管理局 国家卫生健康委员会关于临床急需境外新药审评审批相关事宜的公告》(2018年第79号)规定情形的品种,国家药监局会同国家卫生健康委已组织进行了充分遴选,基本解决了临床急需境外已上市新药在我国上市速度较慢的问题,进一步提高了公众用药的可及性。
2020年,药审中心完成了13个用于治疗罕见病的、临床急需的药品的技术审评,均在规定时限内完成,罕见病药品在3个月之内完成审评,其他临床急需药品在6个月之内完成审评,大大缩短了临床急需境外新药在我国上市的时间差距。截至2020年12月31日,已发布的三批81个品种临床急需境外已上市新药中,已有38家企业的48个品种提出注册申请,其中39个品种已获批上市或完成审评,100%在时限内完成审评工作,详见附件7。
(二)加速推动仿制药一致性评价工作
2020年,药审中心采取切实有效措施加速推进仿制药一致性评价工作。
一是在口服固体制剂一致性评价工作的基础上,积极推进注射剂一致性评价工作。国家药监局于5月12日发布《关于开展化学药品注射剂仿制药质量和疗效一致性评价工作的公告》(2020年第62号),正式启动注射剂一致性评价工作。药审中心发布《化学药品注射剂仿制药质量和疗效一致性评价技术要求》《化学药品注射剂仿制药质量和疗效一致性评价申报资料要求》和《化学药品注射剂(特殊注射剂)仿制药质量和疗效一致性评价技术要求》等技术要求。针对正式启动前已有620件待审评的注射剂一致性评价申请,药审中心成立专项审评工作组,采取细化分类处理措施,严格执行一次性发补,明确注射剂一致性评价注册检查的随机原则,加快审评速度,在不到5个月的时间内完成了620件品种的审评,一致性评价按时限审评已进入常态化。
二是继续规范参比制剂遴选工作,强化服务与指导。药审中心发布《化学仿制药参比制剂遴选申请资料要求》(药审中心通告2020年第32号),进一步强调申请人的自查环节,提高参比制剂遴选工作效率。通过进一步规范内部审核、专家委员会审核等流程,2020年优化了参比制剂遴选工作。自2017年8月开展一致性评价工作以来共发布参比制剂目录35批,涉及3963个品规(1703个品种),其中包括注射剂参比制剂975个品规(405个品种)。
三是加强信息公开和培训。2020年7月举办线上化学仿制药注射剂一致性评价技术研讨会,对注射剂一致性评价技术要求、特殊注射剂技术要求、参比制剂申请资料要求等进行宣讲。
四是持续推进生物等效性试验备案工作。2020年化学药生物等效性试验备案平台共收集了672条记录,仿制药一致性评价生物等效性试验备案平台共收集了292条记录。
(三)全面落实临床试验期间风险管理
为落实《药品管理法》《药品注册管理办法》中有关临床试验期间安全风险管理工作,药审中心在国家药监局指导下,发布了《药物临床试验期间安全信息评估与管理规范(试行)》、《研发期间安全性更新报告管理规范(试行)》和《药物临床试验登记及信息公示管理规范(试行)》3个配套文件。为进一步加强临床试验过程的安全信息监测、识别、评估和风险控制,制定了《药品审评中心药物临床试验期间安全信息评估与风险管理工作程序(试行)》,上线运行“临床试验期间安全风险管理系统”,对临床试验期间的安全信息,如可疑且非预期严重不良反应(SUSAR)和研发期间安全性更新报告(DSUR)等开展全过程信息系统化的风险评估。
2020年药审中心接收来自国内外的SUSAR个例报告164403份(涉及病例为57995例)。其中,来自中国的SUSAR个例报告17243份(涉及病例为4647例);接收DSUR共计1775份;完成临床试验登记2610项(含新冠病毒**和新冠肺炎治疗药物)。对18个药物临床试验中存在的安全性风险,提出了进一步的风险控制处理意见,包括一般风险控制(如修改临床试验方案、修改知情同意书、修改研究者手册、补充完善风险控制措施)和建议申请人主动暂停临床试验等。
面对突如其来的严重新冠肺炎疫情,药审中心探索建立了申请人进行临床试验进展信息报告机制与通道,规范了相关工作程序与技术要求,发布了《新冠肺炎疫情期间药物临床试验管理指导原则(试行)》,制定了规范统一的《应急审批品种临床试验进展和安全监测工作文件》。通过每日和每周的动态风险沟通交流,实施有效的风险监测与风险控制。对2020年2月2日至2020年12月31日经特别审批程序批准15个**、16个生物制品、6个化学药、2个中药的临床试验共39个品种实施动态安全监测,完成了应急审批新冠病毒**及新冠肺炎治疗药物临床试验进展及安全性监测报告共195份。
药审中心参加《药物警戒质量管理规范》(GVP)的起草工作,撰写临床试验期间药物警戒相关内容和要求。完成《临床试验期间安全信息管理:国际医学科学组织理事会(CIOMS)VI工作组报告》的翻译与出版工作,召开“疫情期间临床试验管理及远程智能技术应用学术交流视频会议”,探索开展远程智能化临床试验的安全管理工作,稳步提升临床试验期间安全信息评估和风险管理能力。
(四)继续夯实审评科学基础建设
1.审评质量管理体系建设
发挥审评质量管理体系对药品审评工作持续有效运行的保障作用。一方面是应对新法律规章实施对审评业务工作带来的风险和挑战,结合《药品注册管理办法》及其配套文件要求,及时组织对《质量手册》等体系文件进行全面修订,加强药品审评业务与质量体系的结合;另一方面是应对新冠肺炎疫情对审评工作带来的影响,通过开展药审中心专项内部监督检查,充分锻炼药审中心内审员队伍,及时发现存在的风险并组织改进;同时持续开展年度满意度调查工作,收集国家药监局和申请人对药审中心在落实新注册相关要求、应对新冠肺炎疫情风险防控时的意见和建议,为提高审评质量和效率提供思路,发挥质量体系对各项工作的支持作用。
2.强化审评信息化建设
为确保各项审评改革工作执行过程中的规范化、标准化、数字化,药审中心大力推进信息化建设,依据《药品注册管理办法》和流程为导向的科学管理体系,以药品审评业务流程为基础,立足工作实际,对药品技术审评系统升级完善。新增发补前的专业审评问询和发补后的补充资料问询平台,优化沟通交流系统,加强审评期间与申请人的主动沟通交流,促进审评业务工作开展;新增异议处理审核和注册检验网络通道,调整优先审评审批系统,强化审核流程可操作性,保障审评审批工作顺利实施。开通受理网上预约通道,减少人员流动聚集,有效保障新冠肺炎疫情期间申请人受理业务的有序办理;增加突破性治疗药物程序,为鼓励创新和加快临床急需品种上市拓宽审评通道。通过信息化手段助力药品审评审批业务管理,强化网络信息安全保障,不断提升药品审评审批工作质量和效率。目前药审中心网站申请人之窗实名注册申请企业10674家,基本实现了药品、原料药、辅料、包材注册申请人网上业务办理的全覆盖。
(五)积极推进流程导向科学管理体系建设
为贯彻党的十九届四中、五中全会**,加强治理体系、治理能力建设,以流程导向科学管理体系建设为抓手,不断推进药品审评体系和审评能力的现代化。按照前期工作计划,药审中心已全面铺开任务受理、任务分配、专业审评、综合审评、沟通交流、专家咨询、书面发补、核查检验共8个子课题的科学管理体系试点建设,并印发《药审中心关于运行药品专业审评流程导向科学管理体系有关问题的通知》等8个文件,制定科学管理体系制度制修订计划(含28项制度),截至2020年12月31日已完成14项。注重试点建设成果的信息化,将各项措施纳入审评信息系统,增强措施执行的刚性约束,提高了科学监管和智慧审评能力。
形成按季度汇报机制,定期组织汇报试点运行情况。建立了改革措施管理台账,纳入了58项需要监督的改革措施,按月度对每项改革措施实施的责任落实、进展情况、新问题和解决建议予以一体化动态管理。召开了试点推进座谈会、子课题结题座谈会,对各子课题试点进度、成效、问题等进行总结分析。建立了促进试点建设的长效运行机制,常态化、一体化推进科学审评、高效审评和廉洁审评。
(六)持续开展ICH工作
切实推进我国药品审评审批体系与国际接轨,参与ICH指导原则的国际协调。一是积极参与ICH议题协调工作,自原国家食品药品监督管理总局2017年加入ICH以来,已向36个ICH工作组派出了69名专家,2020年参与ICH工作组电话会437场。二是进一步推进ICH三级指导原则实施工作,国家药监局共发布了3个ICH指导原则适用及推荐适用公告。三是组织开展ICH指导原则培训工作,药审中心开展ICH指导原则线上培训共15场,主要围绕29个ICH指导原则的技术要点、实施现状、实施过程中可能存在的问题等内容进行宣贯。培训对象主要包括国家药监局相关司局、各直属单位、各省级药监局和省级药检机构的相关工作人员,共计2723人观看培训直播,4244人观看直播回放。四是召开ICH指导原则和协调议题研讨会,为广泛听取行业及学界专家意见,2020年药审中心共召开ICH国内专家研讨会15场,共计312人参会。
(七)加强审评队伍建设和管理
加强审评队伍建设,畅通审评员职业发展通道,开展主审审评员认定工作;完善聘期考核评价体系,加强员工聘期考核工作;开展补充性招聘,引进临床、统计等紧缺专业人才;加强员工培训,组织开展《药品注册管理办法》及配套文件系列讲座、实训、英语口语等培训。
九、加强服务指导、改进工作效率和作风
2020年,药审中心驰而不息强化作风建设,积极服务药品高质量发展新要求。
一是紧密围绕新冠肺炎疫情防控大局,超常规创新开展应急审评审批,加强审评服务保障,全力做好新冠病毒**审评过程中的各项工作。面对新冠肺炎疫情对新冠病毒**药物的急迫需求,药审中心坚持尊重科学规律,建立早期介入,持续跟踪,主动服务、研审联动的工作机制,始终保持24小时与企业畅通沟通的状态,无论多晚,即使是凌晨3-4点钟,都会第一时间反馈研发企业诉求,在推动境外临床试验上强化担当,在创新审评审批中挖潜增效,成功高效推动国药集团新冠病毒**附条件批准上市和5个**品种进入Ⅲ期临床试验,确保中国新冠病毒**走在世界前列,及时有力支撑了疫情防控大局。同时贯彻落实习近平总书记坚持中西医结合、中西药并用的重要指示**,主动对接临床救治中应用的“三药三方”生产企业和研发单位,积极做好有效中药方剂转化产品注册和临床试验申请技术指导,确保中药第一时间用于新冠肺炎患者救治。这些成果不仅确保了防疫的应急所需,还为常态化疫情防控准备了重要的战略资源,不仅提振了国人战胜疫情的信心,还为全球疫情防控贡献了中国力量。
二是强化服务申请人沟通交流。在新冠肺炎疫情防控常态化的情况下,全面落实新冠肺炎疫情联防联控措施,最大限度减少人员流动聚集,阻断疫情传播扩散渠道,切实保障申请人的生命安全和身体健康,暂停现场咨询业务的同时开通了电话咨询业务。增设立了8个联系邮箱,申请人可以邮件咨询问题并提供在审品种受理号等信息,项目管理人员将在3个工作日内与该受理号相关的申请人进行联系。通过不断丰富和拓展沟通交流的渠道和方式,服务和便利申请人;为鼓励创新,更好地体现沟通交流的服务属性,结合《药品注册管理办法》,从药物研制规律和注册要求出发,秉持为药品注册申请人服务的原则,修订后发布了《药物研发与技术审评沟通交流管理办法》。在保证受试者安全性的基础上,将Ⅱ类会议划分为依法应沟通交流、原则上应当沟通交流、可以沟通交流三类情形,并明确和细化了三类沟通交流的情形和要求;为提高沟通交流申请办理的可预见性和效率,药审中心通过持续优化沟通交流管理,细化环节时限控制,确保了申请人能够尽快享受到优质的沟通交流服务,努力满足申请人逐年增加的沟通交流需求,将2020年沟通交流申请办理量提升至2019年办理量的1.31倍,这也是2016年办理量的11.35倍。
三是持续改善内部工作作风,提高工作效率。这一年药审中心继续深化审评审批制度改革,持续优化审评流程,严格时限管理,实施审评任务分析会制度,加强项目督导,鼓励药品创新,推动仿制药高质量发展,审评质量和效率有了极大地提升,2020年全年审结任务整体按时完成率创历史新高。药品审评审批的可预期性进一步提高,顺利完成《十三五药品安全规划》涉及药品审评审批改革目标。通过5年来深化药品审评审批制度改革的不懈努力,药审中心实现了量变到质变的飞跃,药品平均审评时限大幅压缩,审评能力大幅提升,一大批创新药、临床急需药获批上市,累计通过和视同通过一致性评价审评的品种已达445个,为十四五药品审评事业的发展奠定了坚实的基础。
药审中心将深入梳理在提高审评效率、创新审评方式等方面的经验做法,使应急状态下的临时性措施,有序地上升为常态化审评工作长效机制,将被动选择但被实践证明长期有效的方法转化为常态化条件下提高审评能力的主动选择。
十、加大药品审评审批信息公开力度
药审中心持续推进技术审评的信息公开工作,提高药品审评审批工作透明度。一是完善信息公开制度,发布了《药品审评审批信息公开管理办法》,明确信息公开的范围、种类、时限等要求,为做好信息公开工作提供了制度依据。二是大力推动新药技术审评报告的公开,自开展工作以来已完成公开新药技术审评报告316个,指导行业研发,更好的服务药品注册申请人和公众。三是加大技术审评过程信息公开力度,通过药审中心网站向申请人进一步公开了审评排队信息、优先审评的状态信息、沟通交流申请及办理信息等信息,新增了“突破性治疗公示”的栏目,公开了“拟突破性治疗品种、异议论证结果”等信息。方便申请人查询信息,进一步拓宽了申请人的沟通渠道,及时回应社会关切,提高了审评审批工作的透明度。
十一、2021年重点工作安排
2020年,药品审评工作取得了一定进展,但仍存在着一些问题:一是注册申请申报量,特别是创新药申报量连年递增,药审中心审评队伍规模结构与审评任务量配比失衡;二是高层次及紧缺专业人才引进难、新进审评员急需长期专业培训等审评能力现代化短板问题突出;三是新旧注册相关规定过渡期间,应及时研究问题,给予相应的解决措施。2021年药审中心将紧密围绕国家药监局工作部署,重点开展以下工作:
(一)积极推动制度体系建设
完善新《药品注册管理办法》配套文件,做好新旧注册相关规定过渡期相关工作,稳妥处理历史问题;继续开展药品审评流程导向科学管理体系建设工作,构建长效运行机制,完善药品技术指导原则体系,规范中心制度体系建设,推动审评体系和审评能力现代化;深入推进监管科学研究,深化与高校、科研院所合作,加快首批重点项目研究成果转化。
(二)毫不放松做好应急审评审批工作
始终保持应急工作状态,完善研审联动机制,坚持特事特办,促进包括中医药、抗体药物等新冠肺炎治疗药物的研发;持续做好应急审评审批核查检验协调工作;继续强化服务指导,持续跟进各条技术路线新冠病毒**研发进展,依法依规严格审评,继续做好新冠肺炎治疗药物和新冠病毒**审评工作;全面总结应急审评审批工作经验,完善审评审批制度体系,进一步激发药品创新发展活力。
(三)加快建立符合中医药特点的中药审评机制体系
构建中医药理论、中药人用经验和临床试验“三结合”的审评证据体系,组建古代经典名方中药复方制剂专家审评委员会,扎实推进中药审评审批改革;参考“三方”审评审批经验,逐步探索适合古代经典名方的中药复方制剂的审评指导原则和标准,完善符合中医药特点的技术指导原则;加快确有临床价值的中药新药审批,发挥中医药在疾病防治中的独特优势。
(四)持续深化审评审批制度改革
巩固按时限审评改革成果,完善项目管理工作机制;完善专家咨询委员会制度,解决争议重大疑难问题,利用巡视整改要求推动制度改革,加大审评审批信息公开力度,优化沟通交流制度,提高审评服务水平;细化上市药品变更管理技术要求,指导药品上市许可持有人开展上市后持续研究;进一步加强临床试验期间安全性评价及药物警戒体系建设;持续推进ICH指导原则在国内转化实施;加快审评数字化建设,推进eCTD系统使用。加快推进研发生产主体信息库建设。
(五)坚持鼓励药品研发创新
持续完善药品审评审批制度体系,坚持以安全有效为根本标准,优化审评资源配置,在创新药审评中探索实施“提前介入”“研审联动”“平行检验”“前置检验”等方式;继续鼓励新药好药研发创新,强化沟通交流,优先配置资源,进一步细化和实施突破性治疗药物、附条件批准、优先审评、特别审批等加快审评程序,加快临床急需境外新药、罕见病用药、儿童用药、重大传染病用药等上市速度。
(六)推动仿制药高质量发展
持续完善仿制药参比制剂遴选,坚持标准不降低的原则,稳妥有序推进仿制药质量和疗效一致性评价;进一步完善仿制药相关技术指导原则和标准体系建设;以建立审评要点体系为基础,推动仿制药审评科学规范、标准,提高仿制药审评质量和效率。
(七)优化人才队伍建设
按照国家药监局统一部署,全力指导和推进长三角、大湾区两个分中心建设;以专业培训为抓手,进一步加强药品审评队伍能力建设;配合药品审评业务,积极开展人员招聘工作,加强队伍建设;进一步加强专业技术队伍建设,完善专业技术队伍晋升等制度;进一步严格人员招聘条件,规范人员离职,严格队伍管理。
十二、结语
大鹏一日同风起,扶摇直上九万里。2021年是实施“十四五”规划的开局之年,药审中心将在国家药监局的坚强领导下,坚持以习近平新时代中国特色社会主义思想为指导,全面贯彻党的十九大和十九届二中、三中、四中、五中全会**,坚持以人民为中心的发展思想,按照立足新发展阶段,贯彻新发展理念,构建新发展格局的要求,以习近平总书记“四个最严”要求为根本遵循,以鼓励创新推动药品高质量发展为主题,以深化药品审评审批制度改革为主线,以满足人民日益增长的美好生活需要为根本目的,以建设国际化现代化科学化药品审评机构为根本动力,坚持为国为民履职尽责,切实保障药品安全有效可及,保护和促进公众健康,努力实现“十四五”时期发展开好局、起好步,以优异成绩迎接中国共产党成立100周年。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2024 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2024 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57






















