https://www.cphi.cn 2018-01-05 11:00 来源:转载 作者:杜丹 杨悦
通过查阅 FDA 官网及有关文献,总结 FDA 仿制药生物等效性研究管理体制。FDA 仿制药生物等效性研究管理体系不断更新完善,在仿制药管理的大整体中,目前 FDA 有较为科学完善的仿制药生物等效性研究的管理体系。我国仿制药生物等效性研究管理起步晚、发展缓慢,仿制药生物等效性研究管理工作的系统化、科学性、专业性均有待提高。我国应建立仿制药申请生物等效性审评专门机构、遵循科学合理的生物等效性审评标准、完善生物等效性研究指导原则、明确生物等效性试验数据资料内容等,促进我国仿制药生物等效性研究管理更加科学与完善。
我国仿制药的质量和安全有效性一直是监管重点,为解决仿制药与原研药之间的差距问题而提出的“仿制药一致性评价”正如火如荼的进行中,同时明确了仿制药体内生物等效是判断仿制药与原研药质量和疗效一致的金标准。生物等效性(bioequivalence,BE)是指,在相似的试验条件及合理试验设计前提下,给予相同剂量的药物等效体或替代体,其活性成分或活性基团在药物作用部位的吸收速度和程度不存在显著差异。
BE的作用是证实等量同种药物的2种制剂生物利用度完全相同,最终使得在替换使用相关的2种制剂时,具有相同的有效性和安全性。目前国际公认BE研究是证明仿制药与原研药生物等效的金标准,本研究针对FDA仿制药BE研究管理的发展情况、当前 BE研究指南体系进行分析介绍,为我国仿制药BE研究管理发展提供借鉴。
1FDA仿制药生物等效性研究管理发展情况
BE的起源
FDA对仿制药生物等效性关注起源于其对药品有效性的关注,可追溯到1962年FDA通过的《Kefauver-Harris Amendment》 药品修订案。该修订案要求,对于1938—1962 年只证明了安全性且已经生效的“告知式”的新药申请,必须补充证明药品的有效性。1962年后FDA又提出了简化新药申请(abbreviated new drug application,ANDA),允许仿制药的申报基于FDA已经通过并公布的原研药的安全性和有效性数据以及通过药效研究实施方案所进行的审评和批准,无需提交全面的临床研究资料,只需证明仿制药与原研药具有生物等效性,由此提出了生物等效性的说法,此为生物等效性研究的萌芽阶段。
BE的地位巩固
科学技术的进步使定量测定生物体液内药物和药物代谢物成为可能,FDA在1965—1976年对仿制药进行了科学评价,1974年FDA生物等效性调查小组发布调查报告表明,在药典标准、GMP指导原则、监管、治疗疗效的差异、制造商、产品质量等方面突出体现了生物等效性的重要程度。基于科学的进步以及鼓励仿制药上市的考虑,同时又不影响新药研发的积极性,1984年FDA通过了《药品价格竞争与专利期补偿法案》,最系统化的提出了生物等效性研究。
BE 的提出基于2个假设,一是生物等效性是药品安全性和有效性的一个良好的替代指标,另一个是在健康人群中取得生物等效性研究数据对患者是等同的。虽然在健康人群中进行的BE研究数据并不能完全等同于患者,但在当时这种假设具有划时代意义。仿制药研发企业不需要再重复原研药企业已进行的Ⅰ~Ⅲ期人体临床安全性和有效性试验,仅需在提交ANDA申请时向FDA提供与参比制剂的生物等效性研究数据。
在批准ANDA时, FDA会同时决定该仿制药是否“治疗等效”于参比品,给予评级, 并公布在《通过等效性评估获得批准的药物名单》(《橙皮书》)中。《药品价格竞争与专利期补偿法案》的提出,创造了仿制药的现代审批体系,同时仿制药生物等效性地位得以确立与巩固。
BE发展完善
1984后BE地位确立,但是当时BE研究理论并不十分完善,监管机构关于生物等效性的详细研究方法、配套指导原则和监管体系尚不全面,BE研究数据造假、用原研样品替换仿制药样品、监管机构人员腐败等问题造成了FDA历史上的仿制药丑闻。仿制药丑闻事件后,FDA在生物等效性实验数据管理方面给予高度重视,强调生物等效性研究的措施包括建立留样要求、经济利益披露要求和增加对从事仿制药生物等效性研究的合同研究组织(CRO)的检查活动,保证了申报数据的准确性。
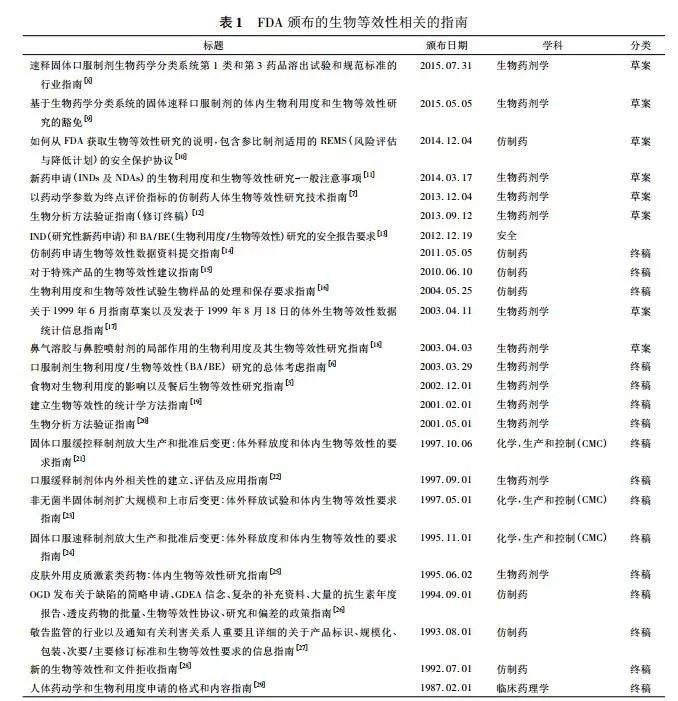
1990年FDA正式成立了仿制药部(OGD),下设2个BE室。为保证仿制药生产商能够顺利完成仿制药生物等效性研究,FDA制定了《药物体内生物等效性试验指导原则》,于1992年7月1日生效,并收载于1995年出版的美国药典(USP)第23版。该指导原则是关于一般药动学和生物等效性资料的统计学分析,主要包括两部分:一部分是关于生物等效性试验的指导原则,另一部分是几个具体药物的生物利用度的实验规程。至此,生物等效性研究便有章可循。随着科学的发展及认知的不断提升,FDA又相继颁布了关于生物等效性研究方法、审评标准、资料数据等方面的多项指南,并发布了很多具体药物BE研究指南,可操作性强,FDA在仿制药BE研究管理方面一直走在世界前列。
2目前FDA仿制药生物等效性受理审批
在美国仿制药申请生产企业可以提交ANDA,施行“一报一批”,申报者可直接进行BE研究,为了避免不必要或不恰当的研究,FDA 建议申请人计划开展BE研究之前,可以提交研究试验方案进行咨询,许多药物也会进行BE预实验,FDA将对研究设计是否恰当 标准物质是否恰当以及拟定化学和分析方法是否充分进行审核,如果合理即可进行,如果存在问题将会给出改进意见,并且FDA接受所有符合GCP的CRO实验数据。
ANDA申请包括申报材料受理和评审2个阶段。申报材料受理时要核实BE研究的完整性,如不完整申请人将会收到“拒收”信,拒收信上将列出BE研究有所遗漏的资料。通过申报受理后进入申报材料评审阶段,此阶段中也包括对BE研究的深入评审。FDA要求申报者提供片剂、胶囊剂、肌内注射剂等具有潜在生物利用度问题的BE试验详细数据资料。
同时FDA也允许申报者对口服液体、静脉注射针剂、滴眼剂等生物利用度明确的剂型进行BE减免。这种情况下,仅申报其仿制药品和参照药的配方和溶解度对比即可。还有些药品,例如透皮制剂,则要求做直接有效性的临床效果对比。
3FDA仿制药申请生物等效性研究指南
FDA有一系列较为细致、具体和严格的关于开展BE研究的指导原则,包括一般性指南和具体药品BE评估指南。
一般性指南
2002年,FDA颁布了《食物对生物利用度的影响以及餐后生物等效性研究指南》。2003年,FDA颁布了《口服制剂生物利用度/生物等效性(BA/BE)研究的总体考虑指南》,且针对具体药物FDA均给出了非常详尽的指导意见。
2013年12月FDA又颁布了《以药动学为终点评价指标的仿制药生物等效性研究指南》,该指南修订并替代了上述2个指南中有关仿制药BE研究的内容。不仅如此,FDA还颁布了许多关于生物等效性方面的指南(详见表1),包括研究方法、生物样品的处理和保存、统计分析、方法验证、数据资料提交、安全、生物等效性研究的豁免等方面,形成了较为完整的仿制药生物等效性研究管理体系。
如果这篇文章侵犯了您的权利,请联系我们。
投稿合作联系方式: Kelly.Xiao@imsinoexpo.com 021-33392297
地址:上海市徐汇区虹桥路355号城开国际大厦7-8楼 200030










