https://www.cphi.cn 2023-12-27 11:42 来源:CPHI制药在线 作者:203的spuer
作为全球药品审评监管程序最为完善以及最为备受瞩目的监管机构,在过去的几年里,尤其是在疫情期间,FDA也正在遭受着前所未有的关注度以及巨大争议,近几年来,FDA有过紧急批准新冠疫苗以及新冠药物,对全球抗疫起到重要的高光时刻,也有过不顾业界质疑仍然宣布批准Biogen的阿尔兹海默病(AD)药物Aduhelm,让FDA公信 力瞬间降到冰点的疑惑操作。
但纵观全球,也很难再找一家和FDA一样,在集全球最顶 尖的法规监管系统的同时却如此饱受争议的监管机构,现如今2023年又将行至尾声,作为疫情后的元年,FDA交出了怎样的答卷呢,答案是截止到圣诞节,FDA CDER(药品评价与研究中心,Center for Drug Evaluation and Research)目前已经批准了55款创新药,在数量上已经远超去年的37款,和2020以及2021年全年批准的数量基本持平,而生物制品评价和研究中心(CBER)批准的基因疗法、细胞疗法、疫苗等数量也超过10款。
在今年,FDA告别了在其任职超过四十年的传奇人物——珍妮特·伍德科克(Janet Woodcock),近40年来,她曾担任FDA代理局长、副局长和CDER主任等职务,宣告了一个时代的落幕。
在告别的同时,FDA在今年也迎来了更多历史性事件,就在本年度,FDA宣布完全批准了继Aduhelm后真正意义上用于治疗阿尔兹海默病的药物Leqembi(lecanemab)、批准了来自大洋彼岸的中国PD-1药物Loqtorzi、批准了首 款CRISPR基因编辑药物Casgevy上市........在FDA已经批准的这55款创新药,就不乏上述充满历史意义的药物,这些药物将在美国甚至是全球范围内起到怎样的作用,又将怎样引领目前的全球医药市场,在未来几年都将会找到答案。
与此同时,根据报道2023年以来FDA累计发出了36份完整回复函(CRL),其中有近一半(47%)来自创新药NDA或者BLA申请,有6款新药在申报新适应症的过程中收到了FDA发出的CRL,此外,有5款改良型新药在申报新剂型、新规格的过程中收到了CRL,而这些药物,将有很大的可能宣布失败,背后的研发企业数十年的心血,也极大可能付之一炬。
有人欢喜有人愁,复杂的情绪夹杂在年尾时刻,但这就是属于FDA,属于新药开发者,属于全球医药的2023年。
01 Leqembi仍饱受争议
Leqembi的故事,想必你我都不陌生,作为同样由卫材/Biogen开发的药物,本年度1月Leqembi获得FDA加速批准并于7月转为完全批准,用于治疗成人阿尔茨海默症,成为自2003年来,首 个获得FDA完全批准的阿尔茨海默症新药。
在有了Aduhelm的前车之鉴,业界以及FDA对于Leqembi的态度则更为谨慎,Leqembi是卫材与Biogen继Aduhelm之后,共同研发的第二款AD新药,但相比之下,不论是临床进展还是获批进展,Leqembi都比Aduhelm更顺利,在2022年9月,卫材与Biogen就宣告Leqembi治疗阿尔兹海默病的III期临床(Clarity AD)达到主要终点和所有次要终点,结果显示,与安慰剂相比,在18个月时,Leqembi组的主要终点CDR-SB评分下降27%,显著的统计学意义支持了后续FDA的审批,也因此让其成为了20年来首 款完全获批的AD新药。
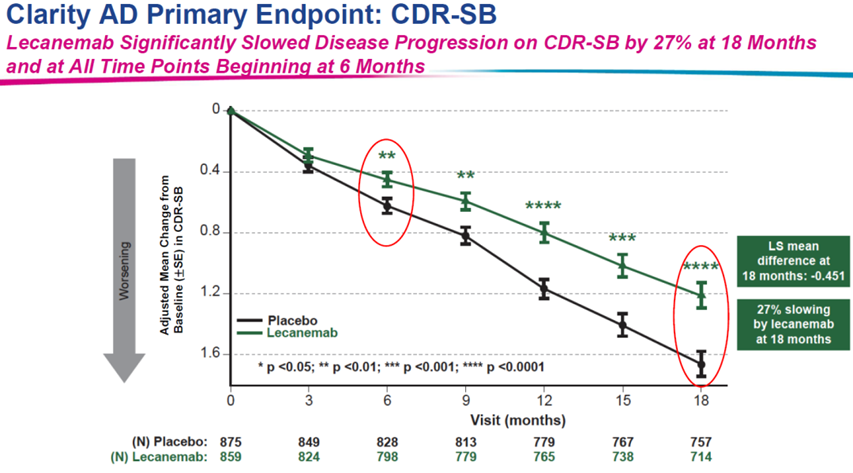
图1:Clarity AD临床试验结果(来自卫材官网)
但Leqembi获批之后却仍旧伴随着巨大争议,首先是研究期间的临床数据表明,接受Leqembi治疗的患者中有 13% 出现脑肿胀,14% 出现脑出血,安全性存在很大隐患,FDA直接在Leqembi 药物标签上给出了黑框警告。
Leqembi的定价为2.65万美元/年,美国医疗保险与医疗补助服务中心(CMS)将在此次完全获批后扩大覆盖范围,进一步促进Leqembi的市场渗透,但此前Aduhelm获批后的巨大争议,商业化失败,证明对AD药物而言,监管机构批准还只是第一步,商业化更是重头戏。根据Biogen的财报,在完全批准后,Leqembi在本年度第三季度的销售额为200万美元。
不过此前卫材发言人透露,预计到2024年3月31日,美国将有约1万名患者使用该药,并预计到2030年,Leqembi的全球年销售额将达到70亿美元。Leqembi可以实现这个商业化目标吗,见仁见智,但对于饱受煎熬的阿尔兹海默病患者而言,FDA的批准给了这些患者一丝曙光。
02 RSV元年开启
呼吸道合胞病毒(RSV)是一种常见的传染性病毒,其属于副黏病毒科肺炎病毒属,可直接影响肺部和呼吸道。据统计,全球RSV年患病率约为3~10%,每年约10.2万名儿童死于RSV感染。此外,老年人亦是RSV易感人群,因年龄所致免疫力下降和基础疾病,老年人患RSV相关严重疾病的风险也很高。
人类RSV疫苗的研发史长达70余年,屡败屡战,足以证明RSV疫苗的开发壁垒极高。但市场前景却非常可观,灼识咨询数据显示,RSV药物全球整体市场规模预计将从2020年的18亿美元增长至2030年的128亿美元,年复合增速达到21.4%。因此RSV药物是各大公司之间的品种之争,也是技术路线之争。
而对于RSV疫苗,本年度FDA接连批准GSK的Arexvy 和辉瑞的Abrysvo则是开启了这一治疗领域元年。Arexvy 于 2023 年 5 月成为首 个获得FDA批准的 RSV 疫苗。Abrysvo于2023 年 6 月获批,是FDA批准的第二款RSV疫苗,获批的时间稍稍晚于Arexvy。
2023年第3季度是两家产品首次正面市场交锋。GSK最新公布的三季度数据显示,Arexvy占据了美国零售疫苗接种量的三分之二,大卖7.09亿欧元,约合8.8亿美元。而Abrysvo则成为辉瑞增长最快的品种,首 个季度销售3.75亿美元,大大超出市场预期。辉瑞正在将Abrysvo 推广至儿科患者群体接种,而GSK则是将目标人群扩大到老年人群体。
03 中国PD-1出海成功
10月27日,Coherus与君实生物宣布,FDA批准其PD-1单抗Loqtorzi(toripalimab,特瑞普利单抗)联合吉西他滨/顺铂作为晚期复发或转移性鼻咽癌患者的一线治疗。其单药也获批用于复发或转移性鼻咽癌含铂治疗后的二线及以上治疗。
鼻咽癌是一种发生于鼻咽部黏膜上皮的恶性肿瘤,是常见的头颈部恶性肿瘤之一。由于原发肿瘤位置的原因,很少采用手术治疗,针对局限性癌症主要采用放疗或放化疗结合进行治疗。
Loqtorzi此次上市申请是基于JUPITER-02及POLARIS-02的研究结果。在JUPITER-02临床III期研究中,与单独化疗相比,Loqtorzi联合化疗显著改善患者的无进展生存期(PFS),将疾病进展或死亡风险降低了48%。该药物还显示总生存期(OS)出现具有统计学显著性和临床意义的改善,与单独化疗相比,特瑞普利单抗导致死亡风险降低37%。
在POLARIS-02临床研究中,Loqtorzi在既往化疗失败的复发性或转移性NPC患者中表现出持久的抗肿瘤活性,客观缓解率(ORR)为20.5%,疾病控制率(DCR)为40.0%,中位OS为17.4个月。
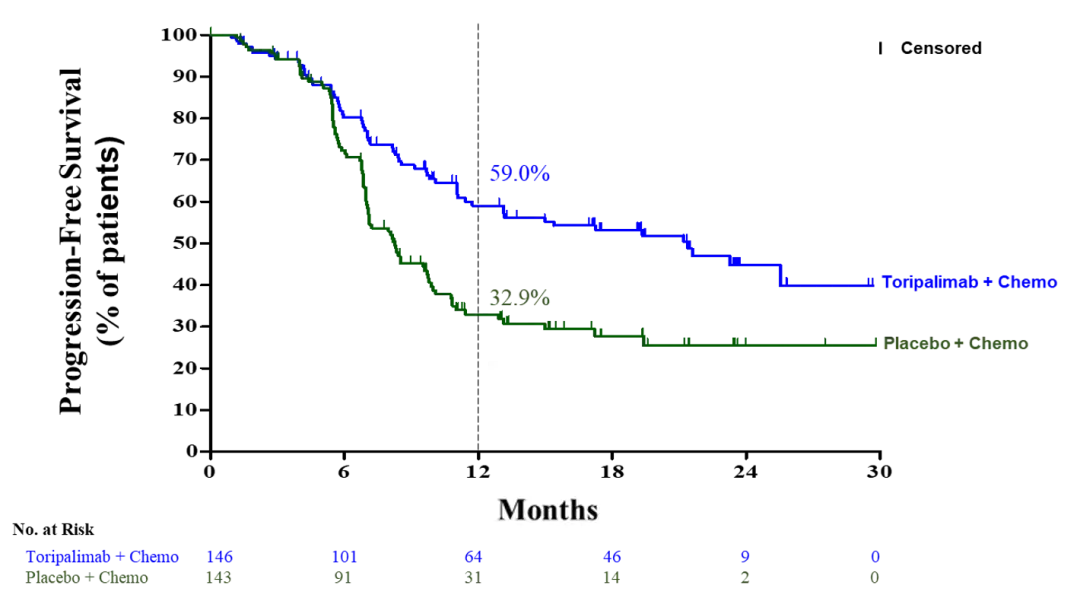
图2:JUPITER-02临床试验结果
Loqtorzi在美国的上市几经周折。由于质控流程变更和现场核查受阻,君实生物曾收到FDA发出的CRL,二次向FDA申报上市后,审评过程中又延期了一次。
目前围绕这款PD-1抑制剂,君实生物的申报适应症已经拓展到食管癌领域,并着手在欧盟和英国进行申请。国内方面,君实生物今年已申报了包括NSCLC围手术期治疗、TNBC一线治疗、RCC一线治疗、ES-SCLC一线治疗4项适应症。
众所周知,中国药企进入美国,尤其是PD-1产品充满了波折。2022年2月,信达生物首 款国产PD-1冲击在美国上市失败。对产业界和投资界信心有了很大冲击。当时,信达生物与合作伙伴礼来采用中国3期临床数据,报批非小细胞肺癌。但是最终结果是,FDA独立的肿瘤药物咨询委员会(ODAC)认为,对于在美国上市的大癌种适应症,需要有美国人群不同民族、种族的临床数据。此外,对于已经有同类产品上市的背景下,临床对照组应选择头对头,并且在临床终点选择上应与美国要求一致。
最终,FDA要求信达/礼来需要补做美国3期临床。而这项3期临床不仅开销巨大,而且耗时将超过3年,不再具有商业化意义,信达生物PD-1出海就此结束。特瑞普利单抗在FDA获批不仅仅是对君实生物的肯定,更是对中国药企在创新药研发领域努力的有力回应。但特瑞普利单抗在美国最终销售情况如何,还得Coherus用实际行动来验证。不过,特瑞普利单抗的获批的确为中国创新药在国际舞台上敲开了一扇门,以君实PD-1与传奇生物CAR-T和百济神州BTK为代表的先驱者,同时也为其他寻求国际认可的中国药企划定了成功的范例。
04 见证基因编辑药物历史时刻
12月8日,FDA批准两款基因疗法上市,分别是Vertex和CRISPR联合推出的CRISPR/Cas9基因编辑疗法exa-cel(商品名:Casgevy)、蓝鸟生物的基因疗法lovo-cel(商品名:Lyfgenia),两款产品均用于治疗伴有复发性血管闭塞危象(VOC)的镰状细胞病(SCD)患者。
Casgevy是全球首 款获批上市的CRISPR基因编辑药物。今年11月中旬,该产品已获英国药品和保健产品监管机构(MHRA)有条件批准上市。Casgevy是一款自体、体外CRISPR/Cas9基因编辑疗法,通过在体外对患者的造血干细胞进行改造,使红细胞可以产生高水平的胎儿血红蛋白(HbF,一种携带氧气的血红蛋白,婴儿出生时自然存在)。因此,exa-cel有可能缓解TDT患者的输血需求,并减少SCD患者的疼痛和血管闭塞性危象(VOC)。
FDA的正式批准也意味着,CRISPR基因编辑疗法获得了全球医药监管标杆的认可,为其后续全球推广奠定了扎实的基础。Casgevy的上市,再一次体现了FDA坚守的原则——引领创新药潮流,解决临床未满足需求。在临床上,尽管CRISPR基因编辑疗法是否存在安全性问题仍有争议,但即便如此,FDA依然顶住压力批准上市,再次释放鼓励创新的信号。
上述几款药物并不能完全代表本年度FDA的成绩,根据统计,在获批新药的分子类型中,小分子药物占比达到55%,显示出蓬勃的生命力。以多肽类和核酸类药物为代表的新分子疗法在过去5年中,占比稳定在12%左右,表明它们已成为重要的新药来源。解决未满足临床需求仍旧是FDA批准药物的黄金法则。
参考资料:
1.FDA官网;
2.医药魔方:2023年,FDA发出36份CRL!临床数据缺乏、CMC缺陷占大头;
3.Pink Sheet:She Didn’t Need To Be Commissioner: Janet Woodcock’s Transformative Legacy
版权所有,未经允许,不得转载。
投稿合作联系方式: Kelly.Xiao@imsinoexpo.com 021-33392297
地址:上海市徐汇区虹桥路355号城开国际大厦7-8楼 200030