https://www.cphi.cn 2024-03-25 14:49 来源:CPHI制药在线 作者:zhulikou431
各国药政部门对制药行业监管,侧重于2个方面。一个方面是对药品品种监管,这类工作主要属于注册类工作;另外一个方面是对生产地址监管。对于生产地址的监管,有的国家或者地区采取许可证制度,例如中国国家药监局就要求生产药品的企业首先要取得药品生产许可证。还有一些国家或者地区,对于药品生产地址监管采取登记制度。不管是上面采取许可证制度的方式,还是采取登记制度的方式,都需要及时掌握某个生产地址的全面信息和动态变化;这样,场地主文件制度(site master file)就应运而生。本文将介绍中国SMF制度的发展情况和技术要求,希望可以为行业提供借鉴。
第一部分:国际SMF法规的情况介绍
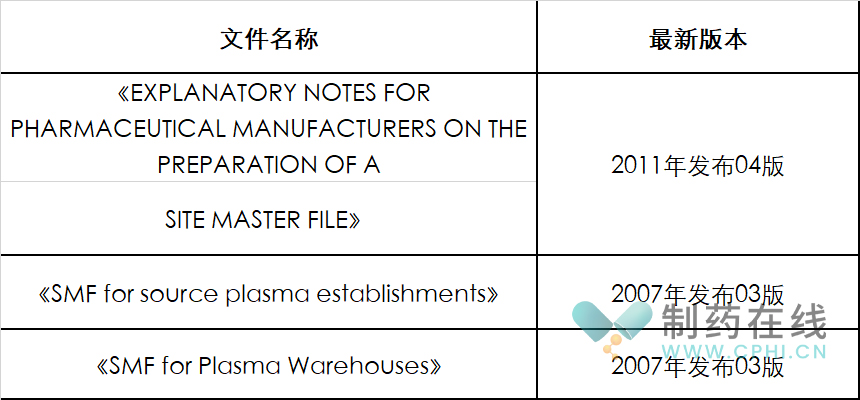
在国际药品认证和注册领域,大部分国家和地区都认可PIC/S组织发布的SMF指南。具体情况汇总如下表:
第二部分:中国SMF的法规依据
《药品生产监督管理办法》2020版规定:
第五条。。。
国家药品监督管理局信息中心负责药品追溯协同服务平台、药品安全信用档案建设和管理,对药品生产场地进行统一编码。
第二十四条从事药品生产活动,应当遵守药品生产质量管理规范,按照国家药品标准、经药品监督管理部门核准的药品注册标准和生产工艺进行生产,按照规定提交并持续更新场地管理文件,对质量体系运行过程进行风险评估和持续改进,保证药品生产全过程持续符合法定要求。生产、检验等记录应当完整准确,不得编造和篡改。
第五十七条监督检查时,药品上市许可持有人和药品生产企业应当根据检查需要说明情况、提供有关材料:
(一)药品生产场地管理文件以及变更材料;
第七十四条 场地管理文件,是指由药品生产企业编写的药品生产活动概述性文件,是药品生产企业质量管理文件体系的一部分。场地管理文件有关要求另行制定。
经批准或者关联审评审批的原料药、辅料和直接接触药品的包装材料和容器生产场地、境外生产场地一并赋予统一编码。
分析:尽管2020年《药品生产监督管理办法》对中国制药企业场地主文件有明确规定,但是由于国家局层面发布的、统一管理的配套文件迟迟未定稿,导致在药品日常监管和注册工作中,很多部门各行其是。就目前现状看,即使在国家局层面,CDE和CFDI也存在各自发布场地主文件的情况。
第三部分:中国SMF文件现状梳理
根据国家局官网和互联网信息,反复检索确认,目前中国药政部门发布的处于有效状态的SMF文件情况如下:
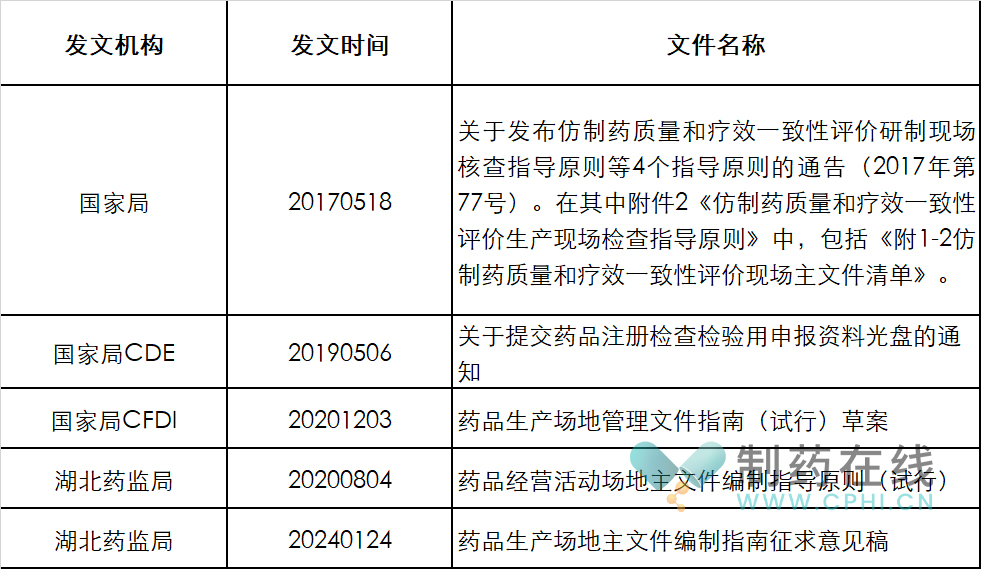
分析:上面这张表展示的信息,详细让中国制药界同仁都觉得有些困惑。对于这份经常使用的重要文件,目前管理现状不能令人满意。
第四部分:中国SMF文件技术要求分析
第一、整体情况
汇总上面表格提到的各机构发布的场地主文件,基本都涵盖如下内容:公司场地总体情况、已经获得批准的生产许可范围、质量体系构成、关键管理团队、厂房生产线布局(需要附图纸)、产品类型、质控方面信息、物料储存信息和其他支持性附件资料。
第二、对于车间生产线布局要求
在这里必须提到一个问题,就是如何划分车间和生产线。国家局目前对此问题无硬性规定,因为不同企业管理风格不同,导致划分的标准差距很大。
在这部分,SMF主要涵盖如下信息:车间和生产线布局图、编号体系、各个车间或者生产线的功能和生产范围、洁净区布局等信息。
第三、对于交叉污染的要求
这部分信息最关键,是SMF文件中的一个主要审核要点。制药企业应该依据2010版GMP通则和2023年3月CFDI发布的共线评估指南来综合评估。
这部分信息和上面车间生产线布局相关,需要结合起来。如果某个生产场地车间和生产线非常多,共线评估内容很多,最好单独附一份评估报告来介绍这部分内容。
第四、 对于自动化系统要求
虽然目前国内发布的多份SMF文件对于自动化系统要求不是很多。然而,随着自动化设备和系统在制药企业各类业务中发挥的作用越来越多,因此企业对于这部分信息要足够重视,最好采用文字和表格的形式来综合描述。
总结
结合上面的信息,可以看到国内各机构发布的场地主文件存在发文机构多、编号不统一、用途不协调的局面。这对于目前正在推动的药政体系改革是不利的,而且也会给制药企业产生很多困扰。由于这项工作拖延很长,导致很多省局在日常对企业监督检查中,并不关注这份重要文件,因此对于一些变更信息的把握,也缺乏敏感性。希望这个混乱的局面尽快结束。
作者简介:zhulikou431,高级工程师、PDA会员、ISPE会员、ECA会员、PQRI会员、资深无菌GMP专家,在无菌工艺开发和验证、药品研发和注册、CTD文件撰写和审核、法规审计、国际认证、国际注册、质量体系建设与维护领域,以及无菌检验、环境监控等领域皆具有较深造诣。近几年开始着力关注制药宏观领域趋势分析和制药企业并购项目的风险管理工作。
相关阅读:
《龙年谈之二:2020-2023年中国企业欧盟认证情况梳理和趋势分析》
版权所有,未经允许,不得转载。
投稿合作联系方式: Kelly.Xiao@imsinoexpo.com 021-33392297
地址:上海市徐汇区虹桥路355号城开国际大厦7-8楼 200030