遗传性视网膜营养不良(IRDS)包括一系列罕见的与导致进行性视网膜退化的基因缺陷相关的疾病。患者从幼年开始到中年,逐渐出现严重的,双侧性的不可逆转的视力丧失。与最常见的IRDS相关的基因缺陷有200多个。美国FDA批准的第一种基因治疗方法,即voreti基因neparvovec(Luxturna; Spark制药),被批准用于成人或儿童RPE65的双等位基因突变引起的IRDS。该疗法于2018年11月获得欧洲药品管理局(EMA)的批准,这些具有里程碑意义的决定将进一步为眼科疾病的基因治疗打开大门。
基因治疗是IRD患者的理想选择,因为病变突变易于发现,并且在某种程度上,眼睛是拥有免疫"特权"的器官:临床试验表明,用于传递所需基因的病毒载体,如腺相关病毒(AAV)和慢病毒,用于眼部疾病治疗时,无明显的免疫反应或系统性不良反应。
最常见的IRD包括色素性视网膜炎、脉络膜病毒血症、Leiber遗传性视神经病变(LHON)、Leiber先天性黑朦(LCA)、Stargardt病、消色差和X连锁视网膜裂(XLRS);在研中的大多数基因治疗是针对这些类型的IRD。视网膜血管疾病和老年性黄斑变性(AMD)的治疗研究也在进行中,尽管这些疾病与单一的基因缺陷无关,在这些适应症中,细胞被基因修饰以产生阻断致病性途径的蛋白质。IRDS基因治疗正在开拓眼科领域产品更广阔的市场。
Luxturna通过视网膜下注射用药,对尚存有活的视网膜细胞的病人发挥治疗作用,因此,病情更为严重的患者不适于Luxturna治疗。虽然有41名患者接受了安全性和有效性评估,但Spark制药仍将进行一项上市后研究来确定该治疗的长期安全性。已知的副作用主要与注射过程本身有关,包括眼部炎症、红肿和疼痛。
行业分析
眼科基因治疗的给药方式取决于靶细胞的位置。在大多数IRDS中,缺陷基因影响视网膜外层、视网膜色素上皮(RPE)和底层绒毛膜层。在这些情况下,病毒载体被传送到视网膜下间隙。LHON治疗时则以视网膜神经节细胞为靶点,将载体注入玻璃体腔内,更好地穿透视网膜内层。同样,在XLRS患者中存在视网膜薄弱,玻璃体内治疗也是治疗XLRS疾病的首选方法。
目前有25种基因治疗处于第I、II或III期阶段(表1)。这些疾病包括一系列眼科疾病,如视网膜色素变性和LCA,以及恶性黑色素瘤和葡萄膜黑色素瘤。应用基因技术公司(AGTC)和梅拉格特公司(MeiraGTx)在眼科基因治疗方面处于领先地位,分别有5项和4项疗法正在开发中。
II期的在研疗法最为激烈,在这个阶段有19种疗法,其中5种用于视网膜色素变性,4种用于色盲,2种用于LCA。
在研的大多数疗法都旨在恢复突变基因的表达(即CHM,编码RAB陪同蛋白1(REP1)、CNGA、CNGB、RPE65、RS1或RPGR)。值得注意的例外包括AMD的治疗,针对该病的病理机制,基因疗法编码一种治疗分子来抑制血管生成或抑制细胞死亡,RST-001基因编码合成通道视紫红质2,该基因具有光敏性。
NightStar的AAV2-REP1,用于治疗脉络膜血症,是目前唯一处于III期阶段的基因疗法,最有望获批上市。NSR-REP1(AAV2-REP1)是一种含有重组人互补DNA的AAV2载体,能在眼部产生REP1。第III期的研究预计将在2020年第一季度完成140名患者的全球招募。
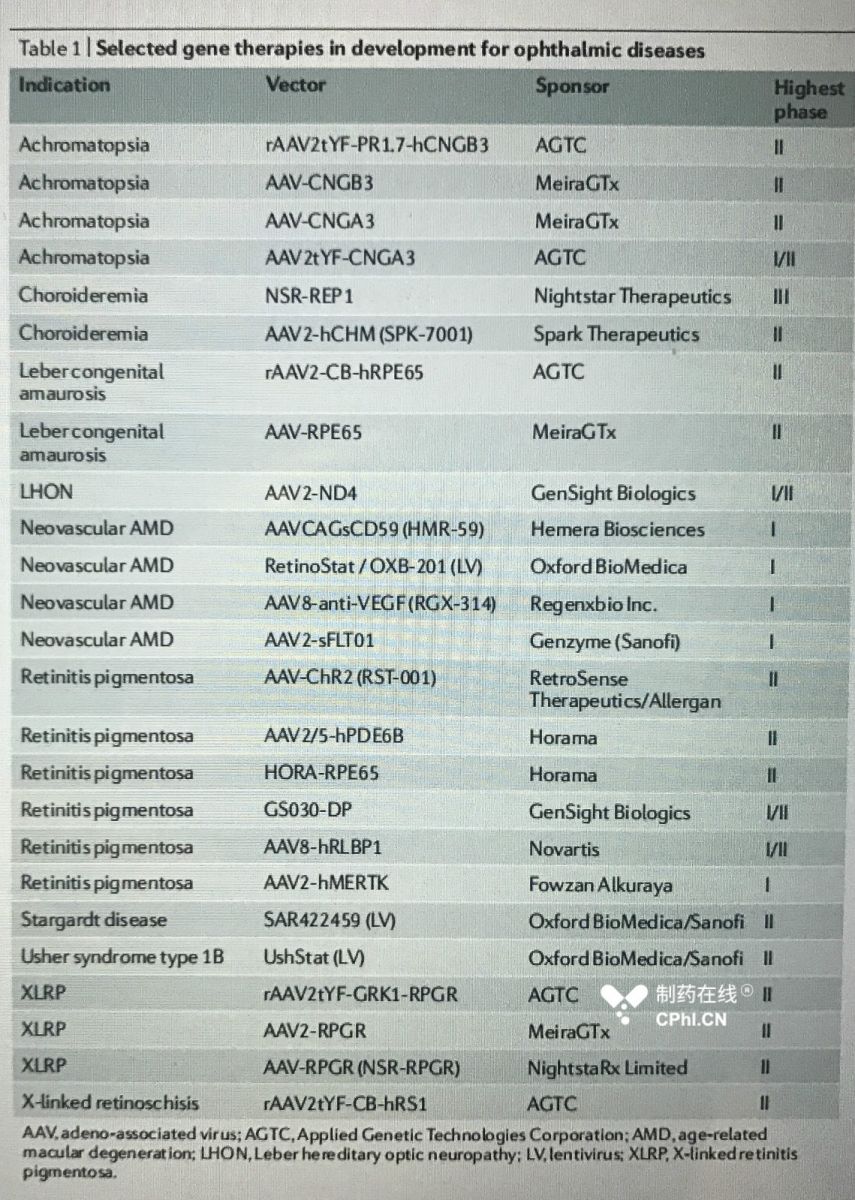
表1
基因治疗当下面临的挑战
虽然第一批基因疗法(Glybera,Imlygic和Luxturna)已经成功地克服了临床发展的障碍,但在生产、临床研究设计、长期安全性研究和未来基因疗法的商业化方面仍然存在巨大的挑战。在各相关研发机构能够从FDA获得试验新药(IND)批准之前,基因治疗需要有良好控制的生产过程和质量验证分析试验,这是由生产周期之间固有的复杂性和潜在可变性决定的。在早期临床前基础研究中,还需要进行更多的生物分布研究,以确保转基因按预期的方式表达,而不被传递到非靶组织。为了帮助克服这些早期发展的挑战,FDA鼓励基因疗法开发人员尽早与其进行沟通,方法是在IND会议前举行一次互动会议,进行有针对性的咨询。
为了避免延误或额外的研究需求,还需要对临床发展进行仔细的规划。基于I、II期研究结果,基因治疗的批准往往会加快;因此,仔细选择研究终点、病人纳入和排除标准以及临床地点选择是至关重要的。对于基于病毒载体的基因治疗,需要筛选抗载体抗体,因为这种抗体会大大限制药物适用人群,而这种限制应纳入成本和时间预测表。
FDA和投资者正越来越多地寻找患者方面的治疗数据,这些数据可以在临床研究过程中获得,作为一个主要的研究终点(比如Luxturna),或者作为补充证据。在持续的基础上获取病人数据,进行长期的安全性随访,可以提供商业战略所需的效率和协同作用。
参考资料:
1. LuxturnaFDAlabel:https://www.fda.gov/downloads/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/UCM589541.pdf
2. Nature Reviews Drug Discovery.



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























