Zolgensma概述
2019年5月24日FDA批准Onasemnogene abeparvovec在美国上市,该药是一种基于腺相关病毒9型载体的基因疗法,用于治疗2岁以下患有运动神经元存活基因1等位突变导致的脊髓性肌萎缩症的儿童患者。商品名为Zolgensma®。
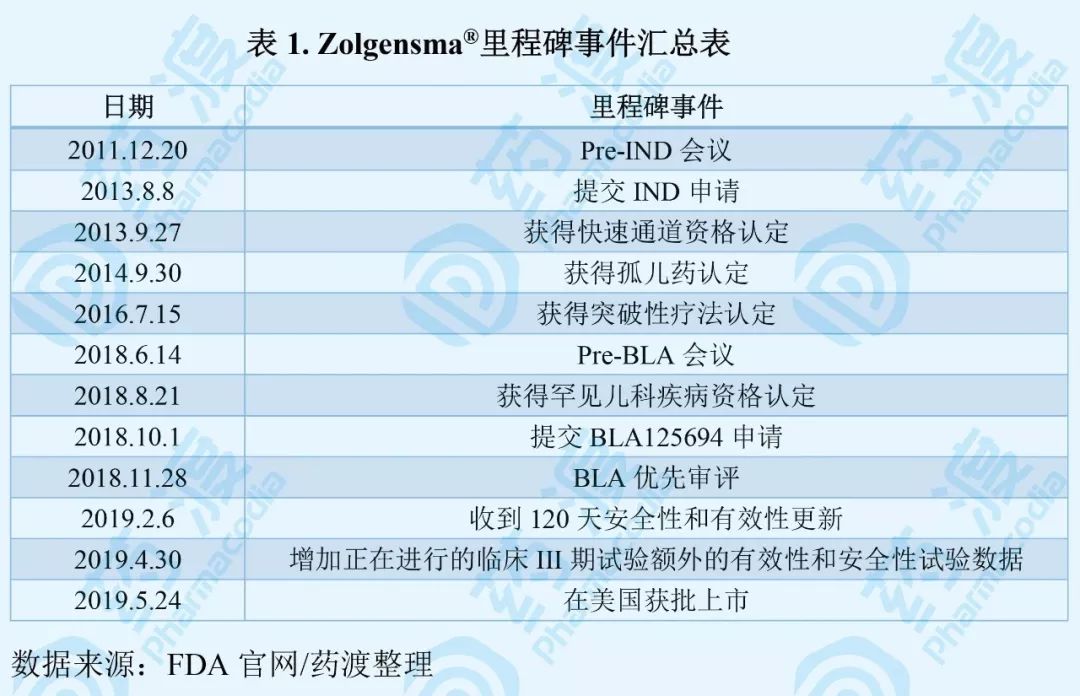
由于临床急需及有望治愈SMN基因1双等位突变导致的SMA患儿,FDA相继授予该药快速通道、突破性疗法、罕见儿科疾病治疗药物、优先审评等相关资格认证,极大的缩短了审评时间,从Zolgensma ®IND申请到FDA批准上市仅历时8年。
Zolgensma®是一种基于腺相关病毒9型(AAV9)载体的基因疗法,腺相关病毒(AAV)载体具有低致病性、对宿主免疫原性弱、长期稳定表达、广泛的细胞和组织特异性强等优点[1]。根据血清型的不同,目前常用的腺相关病毒可分为6型:AAV1、AAV2、AAV5、AAV6、AAV8、AAV9;其中,AAV9较其他病毒载体更容易通过血脑屏障,可以经静脉注射后运输至中枢神经系统,感染约60%的运动神经元[2]。
Novartis/AveXis的Zolgensma®采用Regenxbio公司NAV®基因输送技术平台中的AAV9,利用病毒表面的外壳能结合细胞表面的半乳糖,使得AAV9能穿过血脑屏障,透过血脑屏障把含有SMN基因的AAV9载体递送至中枢神经系统运动神经元。
2018年1月,RegenxBio和AveXis扩大并修订了SMA开发和商业化许可协议。根据协议条款,AveXis将获得Regenxbio公司的NAV®基因递送技术平台开发SMA的全球独家授权。为此,AveXis公司需要向Regenxbio公司支付8000万美元的首付款,一年后额外付款增加3000万美元,两年后额外支付3000万美元,此外还有原始许可协议下的付款和特许权使用费。当Zolgensma®累计销售额达到10亿美元,诺华公司将支付高达8000万美元的潜在商业里程碑付款。
2018年4月,诺华以每股218美元价格收购AveXis,总收购金额高达87亿美元,以此扩大诺华在基因疗法领域的地位。
2018年6月,RegenxBio获得了1亿美元的加速许可证支付,其中包括年费6000万美元和AveXis的4000万美元商业里程碑费。前期的研发投入和高昂的成本注定Zolgensma®价格不菲,诺华推出的定价为Zolgensma®一次性注射治疗费用高达212.5万美元,成为史上最贵药物。
SMA疾病概述
脊髓性肌萎缩症是儿童期较常见的常染色体隐性遗传病,临床主要表现为进行性、对称性四肢和躯干肌肉无力、萎缩,重症患儿呼吸肌常受累、出现呼吸困难、肺炎等症状,常死于呼吸衰竭[3]。该病在欧美人群存活新生儿中发病率为1/10000,携带者频率为1/40-1/50[4]。位居2岁以下儿童致死性遗传病的首位[3]。

SMN靶点概述
研究发现SMA的致病基因——SMN基因定位于5号染色体长臂1区内,SMN蛋白缺失会导致胚胎早期细胞大量凋亡,大部分细胞可以耐受低水平的SMN蛋白,但脊髓前角细胞无法耐受,从而导致SMA疾病[6]。
SMN的修饰基因SMN2与SMN1基因高度同源,SMN1基因缺失或基因内突变造成SMA表型;SMN2基因缺失不造成SMA表型,但其拷贝数量于SMA表型的轻重呈负相关。SMA的主要突变类型为SMN1基因第7或第7、8外显子纯和缺失突变,在我国SMA人群中约占95%。另一种突变类型为复合杂合突变,即1个SMN1基因缺失伴随另1个SMN1基因内的微小突变,约占5%[7]。
SMN1基因存在缺陷而正常的SMN2基因不能完全代偿,最终引起SMN蛋白缺乏是SMA主要的发病机制[7]。
治疗SMA上市药物
SMA最根本的治疗策略是通过各种手段提升具有完整功能的SMN蛋白表达量。治疗手段主要分为依赖SMN治疗和不依赖于SMN治疗。已上市的Spinraza®和Zolgensma®均属于依赖SMN治疗。

Spinraza® Vs. Zolgensma®作用机制
Spinraza®是人工合成的反义寡核苷酸片段,特异性结合于SMN2基因外显子7的剪接抑制序列,促进SMN2外显子7的正确剪接,提升全长SMN2转录的表达,从而增加SMN蛋白表达量。
Zolgensma®是一种基因治疗方法,通过AAV9病毒载体引入人SMN1基因,透过血脑屏障直接递送,直接增加体内SMN蛋白的表达。
Spinraza® Vs. Zolgensma®有效性
FDA根据一项随机、双盲、模拟对照临床III期试验批准Spinraza®上市。在该项试验中,有121名出生不到半年即被诊断出SMA的患儿入组,这些受试者开始接受Spinraza®治疗时也不到7个月大。治疗组和对照组受试患者按2:1随机分组。治疗组接受1次Spinraza®注射(注射到脊髓周围的体液中),而对照组则进行无药物注射(皮肤点刺),观察受试者动作能力(例如头部控制、坐、仰卧位踢腿、翻身、爬行、站立和走路等)的改善情况。121名受试者中有82名的数据可供分析。主要治疗终点为出现运动里程碑反应患者的比例,即Hammersmith婴幼儿神经学检测(HINE)中运动里程碑的改善。与未接受治疗的患者相比,接受Spinraza®治疗的患儿在运动能力(包括全头部控制力、翻身的能力以及独立坐与站的能力)显著改善(40% Vs. 0%, p<0.0001)。今年6月17日,Spinraza®最新说明书中该项试验数据最终更新为(51% Vs. 0%, p<0.0001)。Spinraza®可用于儿童和成人SMA患者使用。
Zolgensma®根据一项正在进行的开放标签、单臂、历史数据对照临床III期试验(STR1VE试验)达到主要治疗终点和一项已完成的临床I期试验(START试验)初步有效性数据获批上市。临床III期试验入组21例婴儿期(出生不到半年)确诊为双等位SMN1缺失和SMN2为2拷贝,且SMN2外显子7缺少c.859G>C修饰的 SMA患者,主要治疗终点设定为1、14个月仍存活的患者比例;2、患儿在18个月大时达到至少30秒无支撑坐立的运动里程碑患者比例。目前正在进一步验证临床III期安全性试验数据。结果显示,相比于对照组,Zolgensma®治疗组14个月患儿存活比例显著提高(67% Vs. 25%);至少30秒 无支撑坐立的运动里程碑患者比例显著增加(47% Vs. 0%)。I期临床试验有效性初步结果为低剂量给药组存活率达到67%、至少30秒无支撑坐立、站立和行走运动里程碑患者比例均为0%;高剂量给药组存活率达到、至少30秒无支撑坐立的运动里程碑患者比例75%、站立和行走患者比例均为16.7%。以上试验结果证实Zolgensma®治疗2岁以下患有SMN1型基因等位突变导致的SMA儿童患者,1型SMA患者。
Spinraza® Vs. Zolgensma®安全性
1型SMA患者使用Spinraza®易出现下呼吸道感染和便秘,2、3、4型SMA患者使用Spinraza®易出现发热、头痛、背痛和呕吐。
Zolgensma®说明书中带有黑框警告,可能发生急性严重肝损伤和转氨酶升高,既往肝功能损害的患者风险较高。在Zolgensma给药前后所有患者需使用全身性皮质类固醇,给药后需要继续检测肝功能3个月。常见不良反应转氨酶升高和呕吐。目前还在进一步获取临床III期安全性数据。
Spinraza® Vs. Zolgensma®给药方式和定价
Spinraza®不能透过血脑屏障,需要重复鞘内给药方式,注射液给药剂量12 mg (5 mL)/次。前4次给药周期如下:前三次给药每次间隔14天。第3次给药30天后进行第4次给药。此后,维持剂量需每4个月给药一次。渤健的定价:Spinraza®首年治疗费用定价为75万美元,此后每年需支付37.5万美元,治疗期通常在5年以上。按以上价格计算5年治疗费用在225万美元,并且每年需要接受治疗,尚不能治愈。
Zolgensma®可以透过血脑屏障,为单次静脉注射给药,一次给药1.1*1014 vg/kg。一次性治疗定价212.5万美元。虽然Zolgensma®单支治疗费用高,但与Spinraza®相比,该药优势在于给予患者一次性注射治疗,治疗效果有望持续终身。并且诺华/AveXis与Accredo®合作,为使用Zolgensma®患者家庭提供长达5年付款计划,以帮助缓解支付方付款压力,按以上付款方式计算,平均每年需要支付42.5万美元,相比于Spinraza®,Zolgensma®略占优势。
Spinraza®Vs. Zolgensma®
从SMA致病机制上分析,SMN1基因存在缺陷而正常的SMN2基因不能完全代偿,最终引起SMN蛋白缺乏时SMA主要的发病机制。Zolgensma®通过替换突变或缺失的SMN1基因来阻止疾病进展,从根本上解决SMA发病机制,有望治愈SMA疾病。而Spinraza®是通过提高SMN2的表达替代缺失的SMN1,通过间接代偿方式治疗SMA。从作用机制上比较,Zolgensma®优于Spinraza®。
从治疗患病人群,目前Spinraza®可以治疗儿童和成人SMA患者,Zolgensma®治疗1型SMA患者,Spinraza®治疗所有类型SMA患者,Spinraza®治疗人群范围更广泛。
从用药安全性分析,Zolgensma®说明书中带有黑框警告,可能发生急性严重肝损伤和转氨酶升高,既往肝功能损害的患者风险较高。在Zolgensma®给药前后所有患者需使用全身性皮质类固醇,给药后需要继续检测肝功能3个月。
从治疗方式分析,Spinraza®不能透过血脑屏障,需要重复鞘内给药,Spinraza®治疗期通常在5年以上,每年需要接受治疗。Zolgensma®可以透过血脑屏障,一次性输注,Zolgensma®优势显著。
从治疗费用分析,Zolgensma®定价史上最贵达到212.5万美元,但通过诺华/AveXis推出的5年分期付款,平均一年治疗费用42.5万美元。Spinraza®首年治疗费用定价为75万美元,此后每年需支付37.5万美元,治疗期通常在5年以上。按以上价格计算5年治疗费用在225万美元。相比较Zolgensma®略占优势。
综合判断,Zolgensma®整体上优于Spinraza®。
根据渤健公司的年报,2016-2018年Spinraza®全球市场销售额分别为0.046亿美元、8.837亿美元和17.242亿美元,增长迅猛。据此估计Zolgensma®全球年销售额有望超过Spinraza®,突破17亿美元。
治疗SMA临床在研药物
进入临床研发阶段的药物主要治疗思路依据SMN基因独特的结构,提高SMN2的表达来代替缺失的SMN1。进入临床阶段有5款药物,具体如下表所示。

在脊髓前角运动神经元发生不可逆的变性损伤之前,对神经肌肉保护措施对SMA患儿运动功能的维持非常重要。不依赖于SMN治疗SMA目前有5款药物进入临床阶段,具体如下表所示。

SMA国内药物治疗现状
2018年8月8日CDE发布的48个境外已上市临床急需新药名单中包括Spinraza®,2018年9月5日渤健联合昆泰向CDE提交国内上市申请,2019年2月25日在中国获批上市,进口获批速度惊人。除了该品种外,罗氏研发的Risdiplam口服溶液用粉末正在国内开展1型,2和3型两项SMA患者安全性和有效性临床II期研究。
目前中国尚无发病率的确切数据,中国人群中的携带者频率约为1/42[9-10]。渤健和罗氏均看好国内SMA市场,但Spinraza®国外的定价很难被国内患者接受,后续在国内上市的定价策略拭目以待。
药渡观点
SMN1基因存在缺陷而正常的SMN2基因不能完全代偿,最终引起SMN蛋白缺乏是导致SMA发病的主要机制。目前,依赖于SMN靶点治疗SMA的有效手段呈现出多样、新颖治疗方式,包括基因治疗、反义寡核苷酸和口服小分子药物等。
Zolgensma®作为基因疗法通过替换突变或缺失的SMN1基因来阻止疾病进展,从根本上解决SMA发病机制,有望治愈SMA疾病。该药能透过血脑屏障,且一次性输注治疗,有望达到治愈疗效。但可能引发急性严重肝损伤和转氨酶升高,既往肝功能损害的患者风险较高。Spinraza®作为一种反义寡核苷酸通过提高SMN2的表达替代缺失的SMN1,通过间接代偿方式治疗SMA。该药不能透过血脑屏障,需要反复鞘内注射,副作用相对较少。小分子药物如组蛋白去乙酰化酶抑制剂或SMN2剪接调节剂等,口服给药,用药方便,且能透过血脑屏障。
SMA由于高致死、致残性,属于严重未满足临床需求疾病,Zolgensma®和Spinraza®申请新药上市均获得FDA给予的快速审评通道、优先审评、孤儿药和罕见儿科疾病治疗药物优先审批等特殊待遇,极大缩短审评时间。Spinraza®作为国内第一批临床急需进口品种,获得CDE的优先审评仅用6个月左右时间在中国获批上市,速度惊人。
全球治疗SMA药物开发热情颇高,呈现出前期小公司投入研发,后期大公司并购公司或收购产品的局面。如诺华高价并购AveXis,将Zolgensma®收入囊中,扩大诺华在基因疗法领域的地位。渤健获得Spinraza®销售权的同时,从AliveGen收购了BIIB110和ALG802,BIIB110与Spinraza®联合使用,可能增强Spinraza®治疗SMA疗效。罗氏收购PTC Therapeutics的Risdiplam和SMN-C3。
以Spinraza®上市三年的市场表现充分说明SMA市场潜力大且存在严重未满足临床需求,Zolgensma®虽然作为史上最贵药物,但随着诺华/AveXis推出5年付款计划和一系列保障计划,上市后市场表现值得期待。若罗氏的小分子口服药Risdiplam获批上市,未来市场将呈现三种不同治疗手段同场竞技,一争高下的局面。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























