伊马替尼的上市开创了靶向药物分子治疗的新领域,但是“道高一尺,魔高一丈”,伊马替尼的反复服用,容易刺激Bcr-Abl激酶发生变异,这也是白血病细胞产生伊马替尼抗性的主要原因,尽管格列卫(伊马替尼)耐药期相对较长,但是,只要患者服用,总有一天会耐药的。
那么,对于治疗耐受伊马替尼的慢性粒细胞白血病(CML)患者怎么办呢?帕纳替尼(ponatinib)将是最好的选择!
帕纳替尼虽然上市晚于伊马替尼10年,却是针对伊马替尼耐药的患者,其研发过程做到了精雕细刻的考察、反复地验证,整个过程是严谨的、是艰辛的,研发的源头性和路径上也具有独立和独创的品格。
1、源于骨质疏松药物的研究
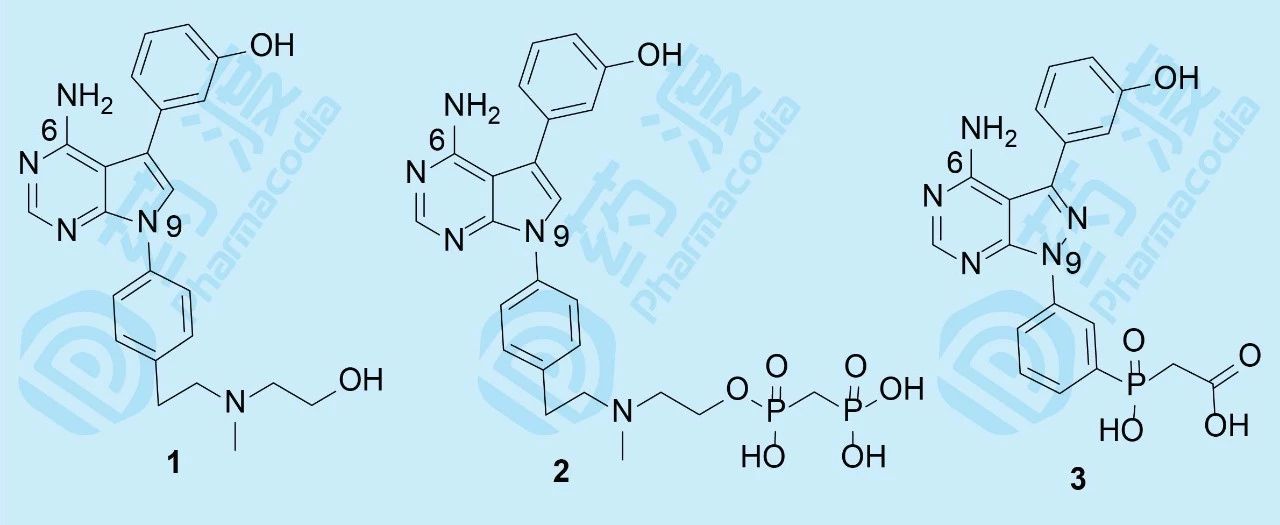
帕纳替尼开始的研究并没有借鉴和模拟伊马替尼的思路,而是源于研究骨质疏松药物。实验证明,Src过表达可引起骨质疏松,Src基因敲除小鼠可避免骨质疏松的发生。基于此,ARIAD公司以Src激酶为靶标研制抗骨质疏松药物。选择嘧啶并吡咯和嘧啶并吡唑类化合物作为筛选苗头,源于诺华和辉瑞公司曾报道过这类化合物具有活性。ARIAD发现化合物1抑制Src激酶IC50值40 nmol·L-1, 进而变换结构,化合物2和3的IC50值分别为4 nmol·L-1和20 nmol·L-1。
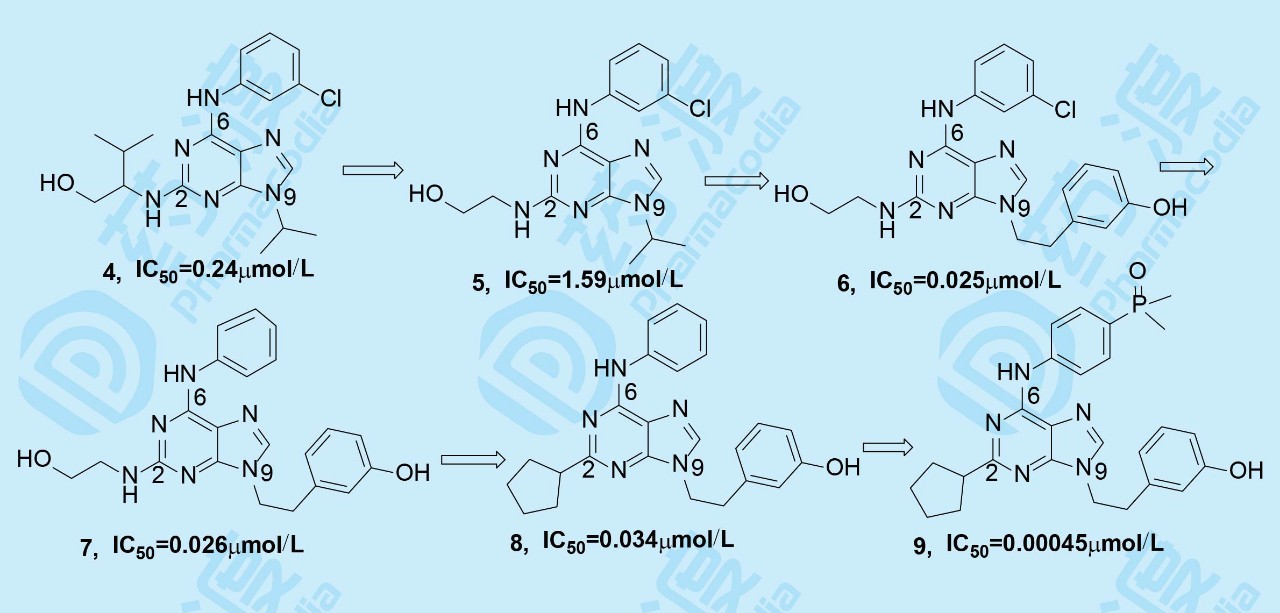
在研究上述内容的同时,ARIAD还启动了以依赖周期蛋白激酶(CDK)为靶标,研发抗骨质疏松的课题。2,6,9-三取代嘌呤化合物purvalanol A(4)是CDK强效抑制剂。化合物4抑制CDK2的活性IC50高达70nmol·L-1,但抑制与骨质疏松相关的Src激酶活性不高,IC50=0.24μmol·L-1,由此设计合成了在嘌呤2,6,9-位变换不同取代的化合物。
将化合物4中2位的异丙基去除后,活性降低6倍;再将9位的异丙基换成间羟基苯乙基,活性提高60倍,这提示9位为大片段有利于同靶标的结合。6位的氯苯基去除氯原子后活性几乎不变,但是2位烷胺基换成环戊基时活性略有下降,而6位的苯环对位连接氧化膦基或亚甲二膦酸基时,对Src激酶抑制活性显著提高,其中化合物9活性最高,IC50=0.45nmol·L-1,代号为AP23464,为里程碑式化合物。
2、转向研究CML的双重抑制剂
持续服用伊马替尼引起的耐药性是由于激酶的突变,除了Abl变异之外,Src 酪氨酸激酶家族的Lyn、Hck和Lyn等也发生突变, ARIAD进而以耐药的CML为目标,研究对Src/Abl双靶标抑制的药物。
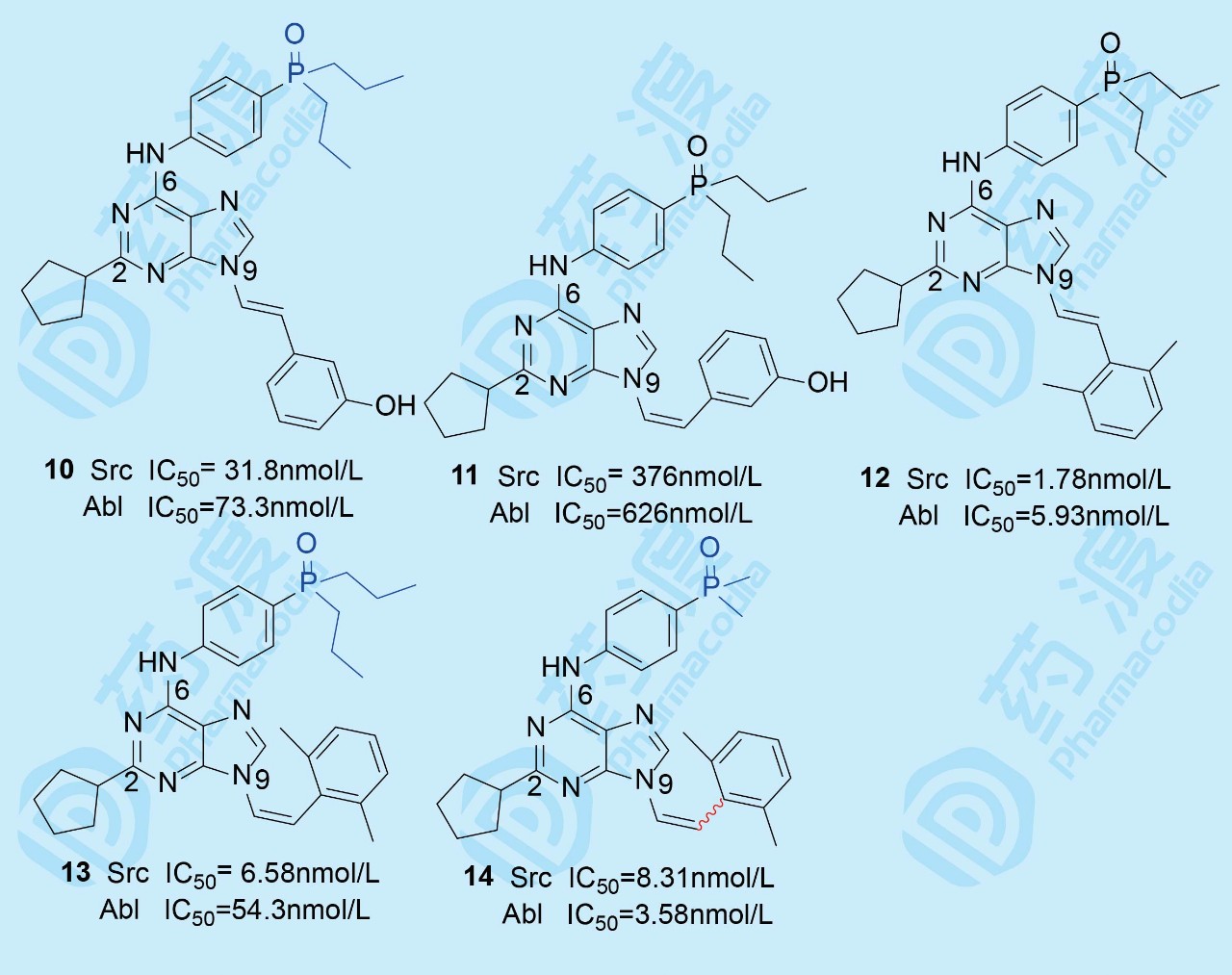
化合物9对两个激酶Src/Abl的抑制作用IC50都低于1 nmol·L-1, 根据化合物9与Src晶体结构信息,认为将亚乙基的柔性连接基刚性化,以固定末端苯环的取向,应能提高活性。用分子对接方法将N9连接的乙苯基变换成trans-乙烯苯10,由于双键的定向作用,使苯基结合于激酶突变后产生的特异性疏水腔。同时也合成了cis-乙烯苯11以验证对接结果。进而模仿达沙替尼的结构片段,合成了trans-乙烯2,6-二甲苯基化合物12和cis-乙烯2,6二甲苯基13,抑制酶和细胞的活性进一步提高。然而,化合物10和12对Src和Abl抑制活性相差较大, 为提高对Abl激酶的活性,将二丙膦氧基片段换为体积较小的二甲基化合物14,拉近了对Src和Abl的活性,为此固定二甲基膦氧基不变,变换N9乙烯基连接的芳环。
3、另一路径——设计DFG-out构象的抑制剂
上述以化合物9为先导物所优化的双重抑制剂,N9连接的片段所结合的位点是活化环套的DFG呈与ATP结合的状态,即结合于DFG-in构象的抑制剂。同时进行的另一途径是合成激酶的活化环套呈DFG-out构象的抑制剂,犹如伊马替尼结合样式。Abl激酶与抑制剂的晶体结构表明,当活化环套呈DFG-out构象时,抑制剂N9连接的二芳酰胺片段与酶形成了氢键网络,故而设计了化合物15。
3.1 C6取代基的变换
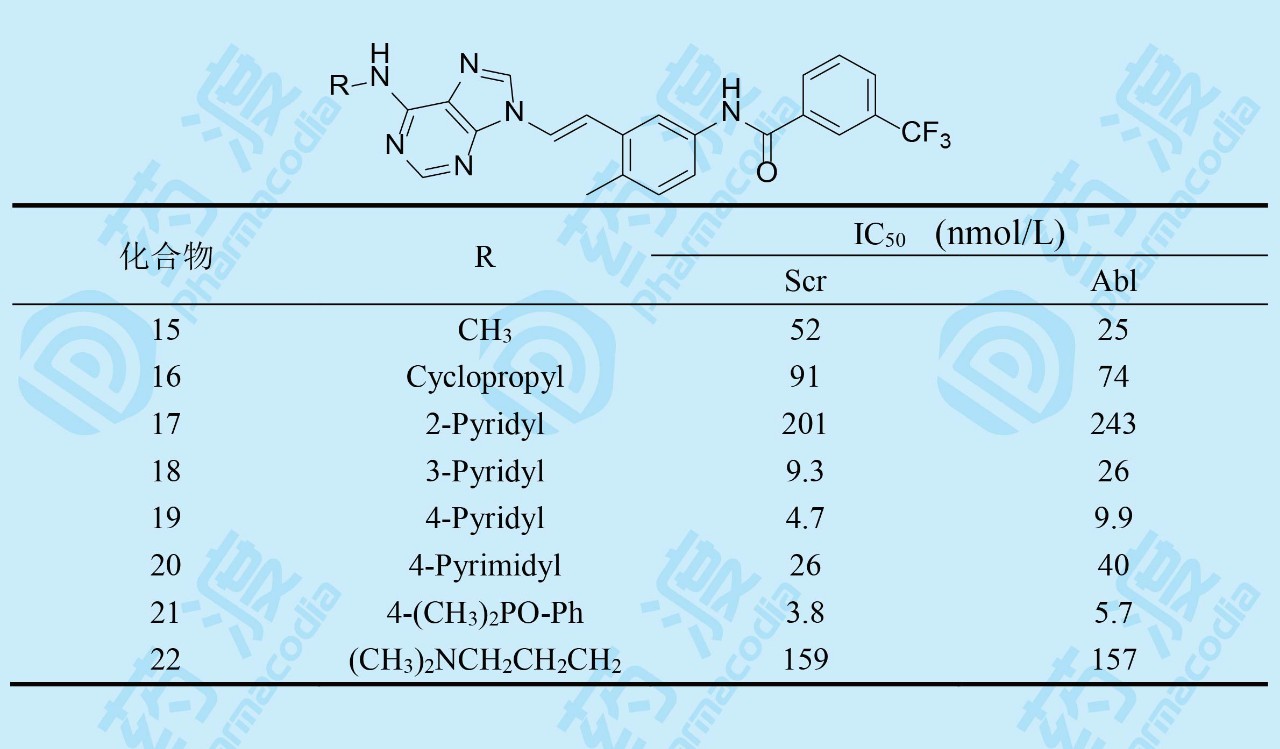
活性评价结果证实,化合物15对Src和Abl都有较高活性,而且在10 μmol·L-1浓度下对正常细胞无作用,提示有高选择性。大鼠药代动力学实验表明15的口服利用度F=20%,半衰期很短,是由于C6的甲胺基发生氧化去甲基作用。故而合成了环丙基化合物16(环丙基代谢稳定性高于甲基),仍保持了酶和细胞活性,而改善了药代,F=40%,血浆中有高暴露量。环丙基的亲脂性强于甲基,推论进一步增高亲脂性会有利于活性。为此合成了芳环17~22,吡啶和嘧啶化合物活性都很高,但2-吡啶基化合物17却很低,可能是由于2'-N与嘌呤N1的电性排斥作用,芳环扭转,破坏了共面性,而18和19没有排斥作用(但嘧啶也有邻位排斥作用,活性下降却不明显)。化合物21移植了9的模块,Scr和Abl活性都比较高。22的活性很弱。然而这些高活性化合物的大鼠药代都不好,因而将C6-环丙胺基固定做进一步优化。
3.2 N9连接片段的变换——借鉴尼洛替尼的设计
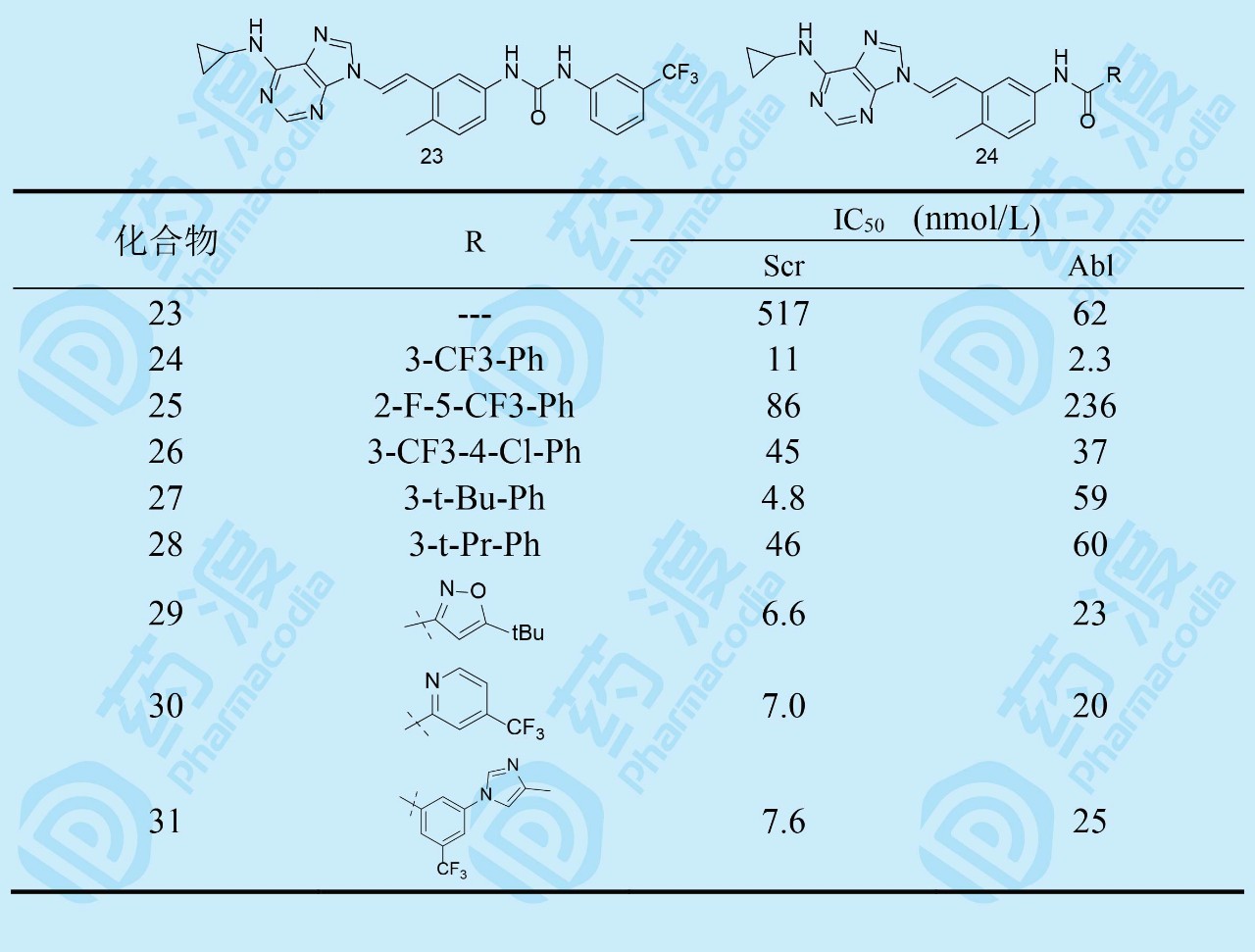
优化N9位置,借鉴了诺华的尼洛替尼的设计理念,合成了脲基和反向酰胺连接的化合物23和24。23几乎没有活性,而24的-CONH-比区域异构体16的-NHCO-活性强,犹如尼洛替尼活性强于伊马替尼。为此,以24为新起点,优化末端芳环。构效关系表明,在三氟甲苯基环上再加入卤素(25、26)活性减弱,三氟甲基换成叔丁基(27),对Src活性提高一倍多,但抑制Abl作用降低,提示这两种激酶结合腔的差异。化合物28为异丙基,都比叔丁基弱。然而27亲脂性过强,clogP=6.76,超过化合物24,综合考虑27不可取。含叔丁基的芳杂环化合物中异噁唑29对Scr和Abl都很强,但大鼠药代不如24。化合物30的活性优于24,而且亲脂性比24降低10倍。有趣的是,将上市药物尼洛替尼的“大块片段”连接在酰胺上,化合物31活性非常高,说明该优化的模块适用于本系列中,也意味着与激酶结合的相似性。
3.3 连接基乙烯基的变换
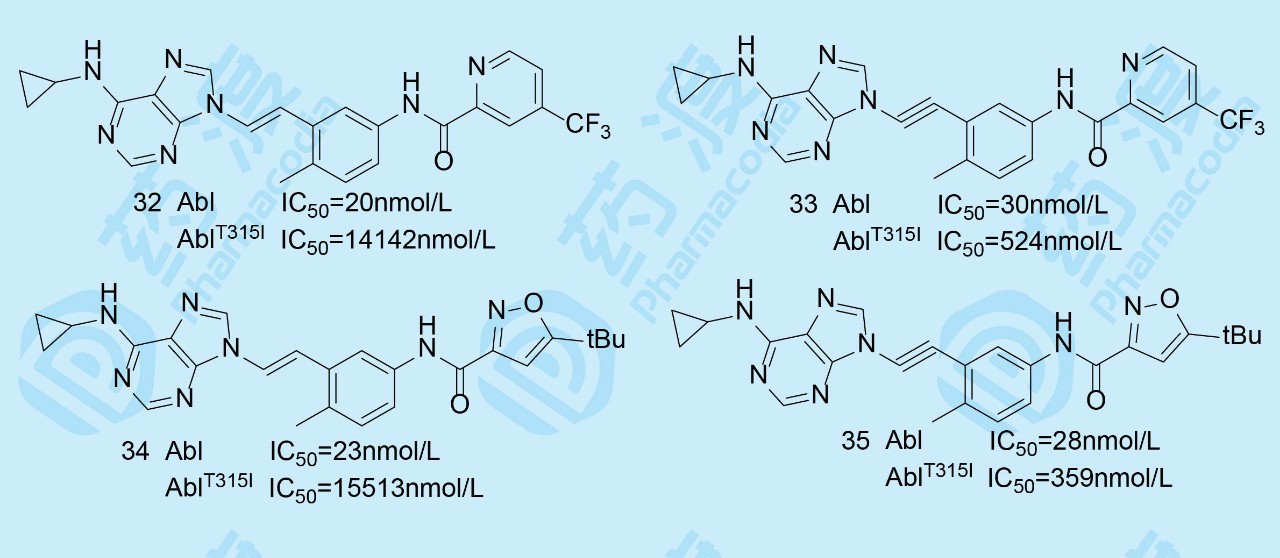
持续应用伊马替尼可引起Abl激酶发生变异(AblT315I),即门户残基Thr315 突变Ile315,T315I突变的后果。氨基酸侧链由CH(CH3)OH变成CH(CH3)CH2CH3,不仅体积变大,空间上也阻碍了伊马替尼的进入,而且失去形成氢键的能力(羟基O为氢键接受体)。化合物31对AblT315I激酶有中等强度的活性,提示该系列化合物的结构特征对门户残基的Abl变异仍可结合。进而设想将乙烯基换成直线型的乙炔基更能解除位阻效应。为此将乙烯基和乙炔基与有强结合作用的芳环进行组合优化。
化合物33与32比较(同样35与34相比)虽然对未变异的Abl的活性相近,但对变异的激酶活性(AblT315I)提高了30~40倍,这些结果说明乙炔基是优化的连接基。然而化合物33和35的药代动力学性质不佳,且很容易被代谢消除,须进一步优化。
3.4 骨架迁越——更换嘌呤环
嘌呤和苯环之间的连接基是N-C≡C-C的炔胺结构时,化学和药动学不如C-C≡C-C稳定,故试图变换嘌呤环结构,合成一系列新的化合物。
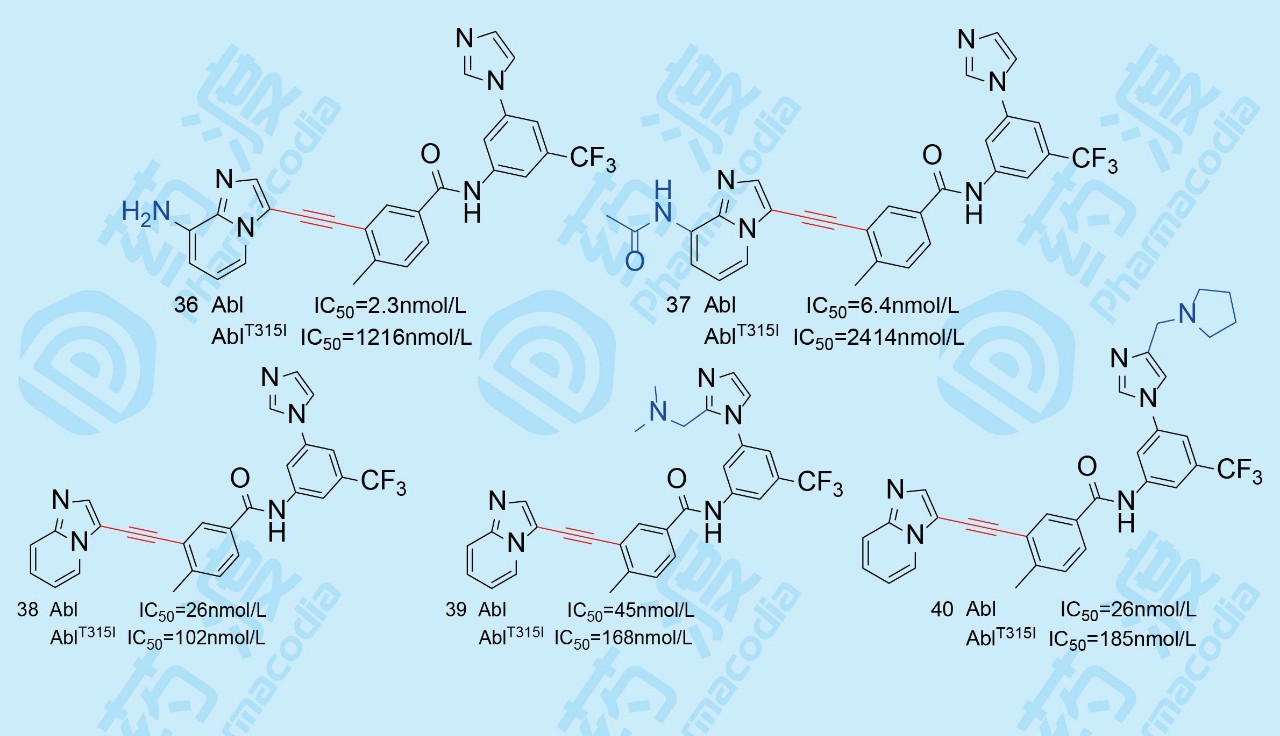
化合物36和37对Abl的抑制活性很高,证明了骨架跃迁的成功,但是大鼠的药代动力学不佳,可能是由于嘌呤环上的氨基或乙酰氨基导致的,故去掉氨基后,化合物38保持了对变异酶的活性,而且改善了大鼠药代,口服利用度42%。但Abl的活性弱于36和37。在咪唑环上加入助溶基团后,39和40的活性仍不高,还得以新的途径优化活性。
3.5增加与活化环套的结合——模拟伊马替尼的结构设计
在已有优化的基础上再提高与AblT315I酶的结合作用,探索增加与活化环套(A-loop)的残基结合。
合成化合物41的活性结果表明,对AblT315I激酶活性比38提高2倍,Abl提高20倍,而且在高浓度(340 μmol·L-1)人血浆白蛋白存在下对白血病细胞抑制活性并不减弱,提示与血浆蛋白结合的较少,因而成为一个很好的突破口。若将哌嗪片段移至5位,活性减弱,提示位置的重要性。下一步优化碱性片段都在苯环的4位。
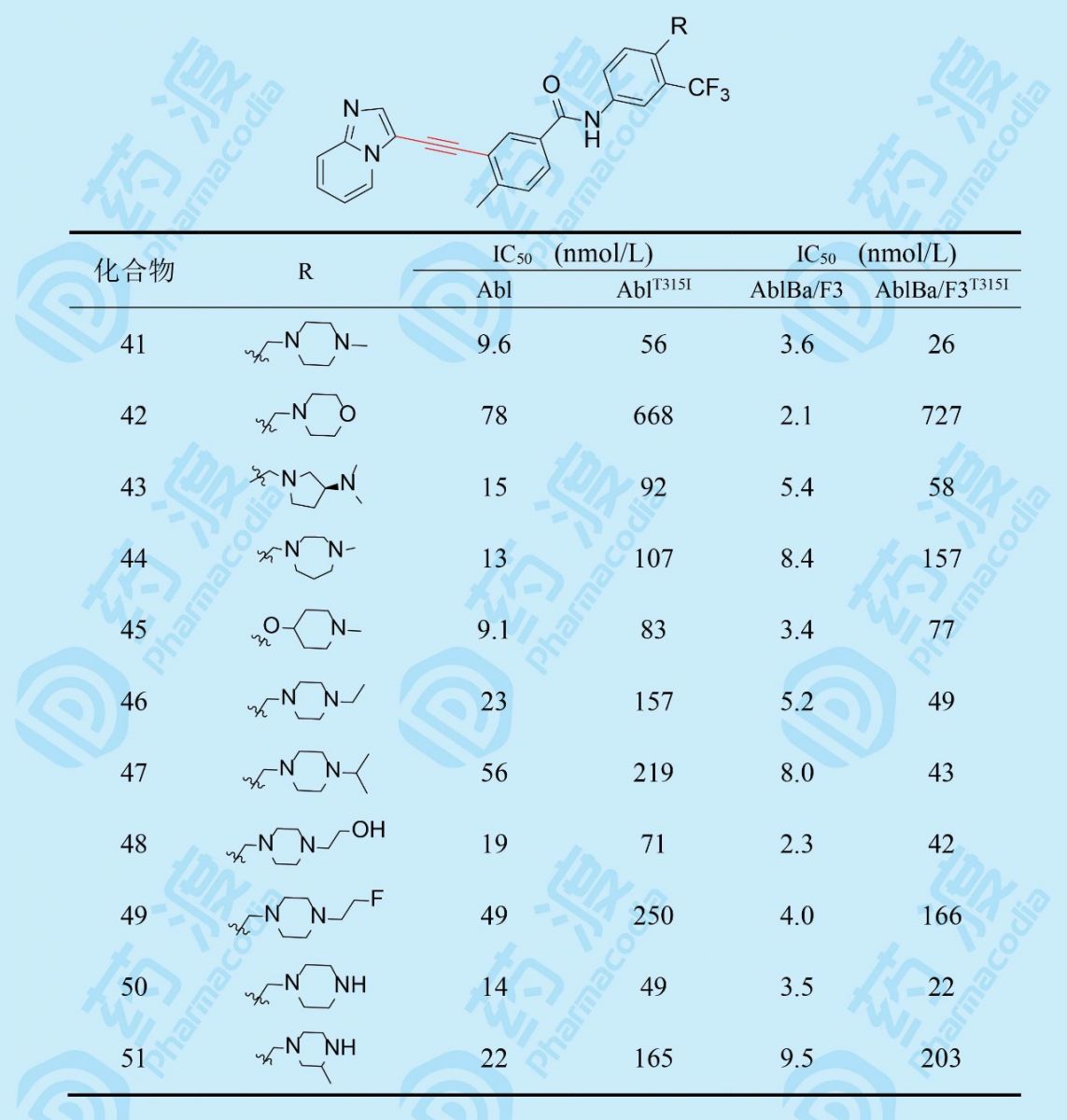
变换碱基片段的活性结果表明,化合物41的哌嗪N4被氧原子替换,吗啉化合物42活性显著减弱,佐证了N4形成氢键的重要性。43和44分别为缩环和扩环化合物,虽然也存在类似于哌嗪N4的氮原子,但这两个化合物的活性弱于41,将41的N-CH3换成较大的烷基或氟(或羟)乙基(48、49)都不利于抑制突变株。
3.6母核吡咯并吡啶的再检讨
当初去除氨基以改善药代效果时,虽然损失了一些活性(权衡结果划算),现为了弥补损失,合成了一系列增加杂环内的NH的衍生物,以提供氢键给体。
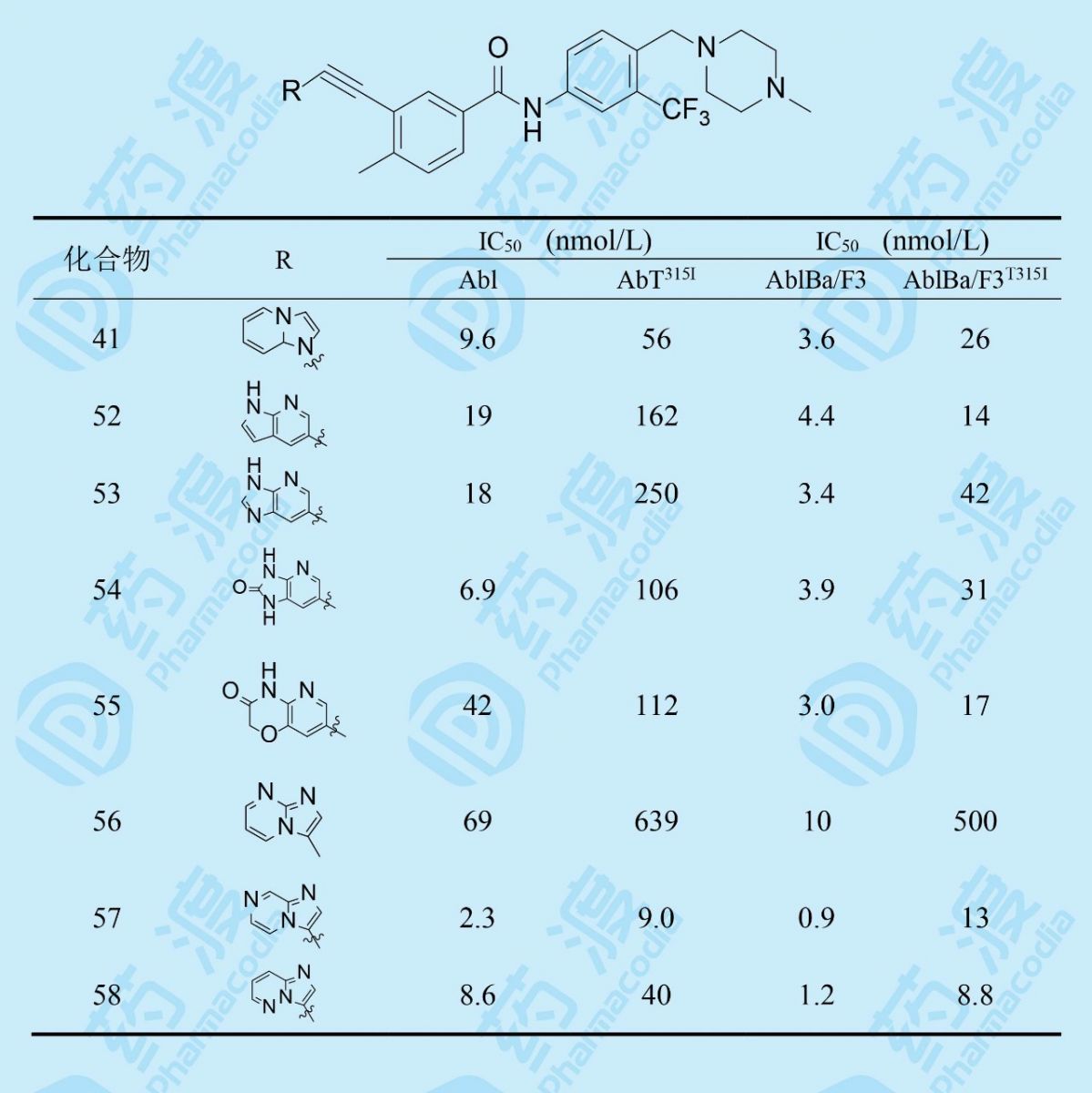
化合物52~55的活性表明,52和55对变异激酶的活性略有提高,但是53和54的活性降低。另一思路是,为了降低41的亲脂性(clogP=6.69),在吡唑并吡啶环的不同位置加入一个氮原子(加一氮原子分配系数降低1个对数单位),化合物57和58活性显著提高,尤其是57。56的低活性是由于杂环的8位是用氢键接受体占据了氢键给体的位置,从而发生互相排斥的缘故。58的大鼠药代优于57,下一步是对母核换作咪唑并吡啶后的继续优化。
3.7 新骨架的各取代基的综合调整
以58为新一轮优化的起点,在苯环A的2位变换甲基,苯环B的3,4位变换三氟甲基和碱基,对结构进行微调,化合物及其活性如下:
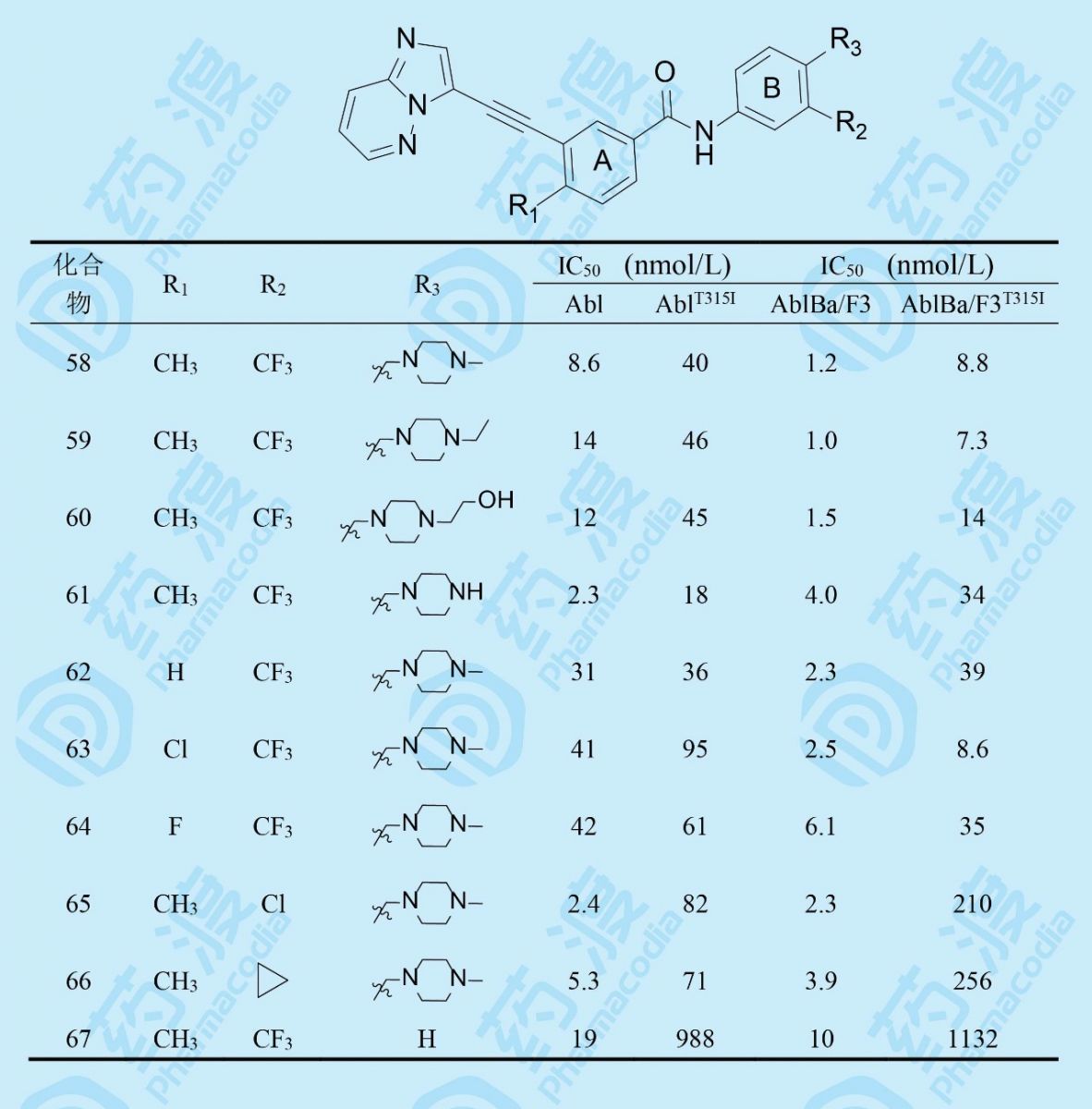
得出还是化合物58的活性最佳。
3.8候选化合物的药代性质
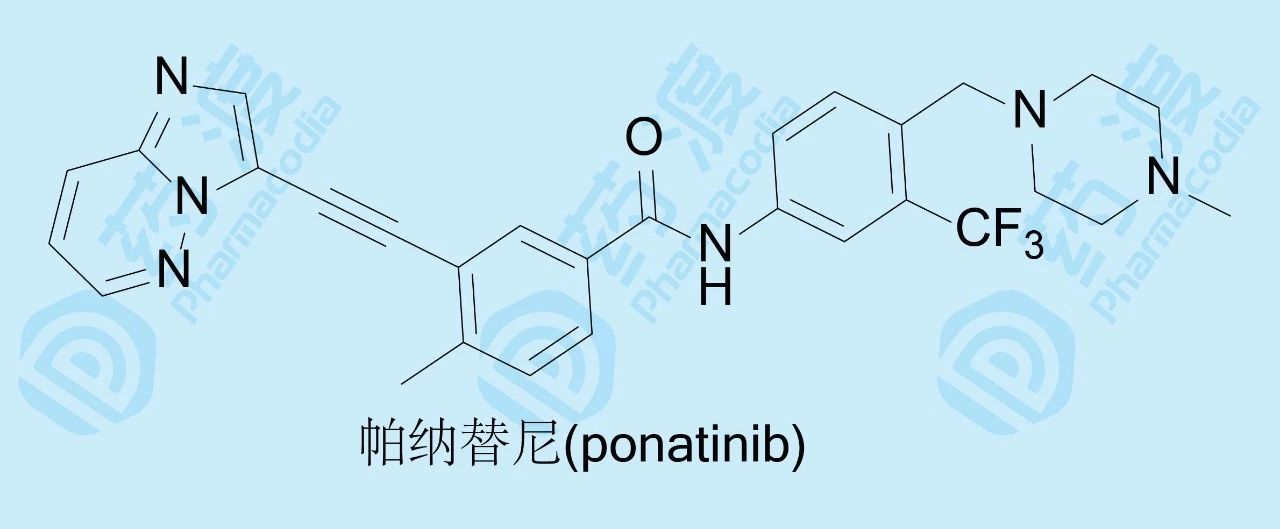
化合物58胜出为候选化合物,与各个里程碑化合物作了药代动力学比较,58大鼠灌胃的生物利用度18%,半衰期11h,静脉注射的清除率0.65 L·h-1·kg-1。58定名为帕纳替尼(ponatinib),临床前和临床实验,表明对慢性粒细胞白血病和费城染色体阳性的急性白血病有效,于2012年经美国FDA批准上市。
小结
帕纳替尼是一个线型分子,它的研制涉及了整个分子所有的基团和片段的优化,虽然同类产品伊马替尼在它10年前面市,也在设计上有所借鉴,但仍不失为创新性药物。在分子设计?合成?评价?构效关系分析的循环反馈中,结构生物学、分子模拟和药物化学的交织应用与印证,对结构的各个侧面做到了精雕细刻的考察,甚至是反复地验证。
参考资料
1. Wang Y, Metcalf CA, Shakespeare WC, et al. Bonetargeted2,6,9 -trisubstituted purines: novel inhibitors of Src tyrosine kinase for the treatment of bone diseases. Bioorg Med Chem Lett, 2003, 13: 3067-3070
2. Sundaramoorthi R, Shakespeare WC, Keenan TP, et al. Bonetargeted Src kinase inhibitors: novel pyrrolo-and pyrazolopyrimidine analogues. Bioorg Med Chem Lett, 2003, 13: 3063-3066
3. Wang W, Metcalf CA, III, Shakespeare WC, et al. Bonetargeted 2, 6, 9-trisubstituted purines: novel inhibitors of Src tyrosine kinase for the treatment of bone diseases. Bioorg Med Chem Lett, 2003, 13: 3067-3070
4. Huang WS, Zhu X, WangY, et al. 9-(Arenethenyl) purines as dual Src/ABL kinaseinhibitors targeting the inactive conformation: design, synthesis, and biological evaluation. J Med Chem, 2009,52: 4743-4756
5. Huang WS, Metcalf CA, Sundaramoorthi R, et al. Discovery of 3- [2- (imidazo[1,2-b] pyridazin-3-yl)ethynyl]-4-methyl-N-{4-[(4-methylpiperazin-1-yl)- methyl]-3-(trifluoromethyl)phenyl}benzamide(AP24534), a potent, orally active pan-inhibitor of breakpoint cluster region -abelson (BCR - ABL) kinase including the T315I gatekeeper mutant. J Med Chem, 2010, 53: 4701-4719



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























