辉瑞是率先专注JAK抑制剂的制药巨头,其有完备的JAK抑制剂研发管线,并且成功推出了全球首个上市的JAK抑制剂托法替尼。2018年9月,辉瑞公司宣布,其研发的口服Janus激酶3(JAK3)抑制剂PF-06651600用于治疗斑秃获得了美国食品和药物管理局(FDA)突破性疗法的认定(breakthroughtherapy designation),为全球斑秃患者带来新的曙光。目前,PF-06651600治疗斑秃的Phase III 临床实验(NCT04006457, NCT03732807)正在进行当中。随着临床实验的进展,实验室内制备PF-06651600的初始路线已不能满足临床实验API的需求。为实现大规模,快速,稳定地制备临床实验所需API原料,新的工艺路线亟待开发。
PF-06651600初始合成路线
如图一所示,PF-06651600初始合成路线以6-甲基3-胺基吡啶(2)作为起始物料,经Boc保护得到化合物3。而后,以贵金属PtO2作为催化剂,在55 psi H2下,于50°C乙酸溶液中完成氢化,得到外消旋哌 啶rac-4,顺式/反式比例为2/1。将rac-4哌 啶N原子以Cbz保护基进行保护,分别得到2∶1的顺式rac-5和反式rac-6混合物。接着,以手性超临界流体色谱法(SFC)对该非对映异构体混合物进行分离,得到顺式rac-5。将获得的rac-5在二氯甲烷溶液中以HCl除去N-Boc基团,得到盐酸盐形式的 rac-7。rac-7·HCl与2,4二氯吡咯并嘧啶8之间进行亲核取代反应得到中间体rac-9。随后9在Pd/C催化下进行氢化,同时除去N-Cbz保护基和2-氯取代基,得到rac-10。在Schotten-Baumann条件下用丙烯酰氯与rac-10反应,粗产物经硅胶柱层析纯化得到目标化合物消旋体(rac-1)。最终,经再一次手性SFC分离得到光学纯的1。辉瑞研发人员通过该路线共提供了约60 g 目标产物1(纯度约95%,ee≥98%)。该路线最大产量为40 g,总收率为5%,可支持生物活性测定,ADME评估和早期毒理学研究。
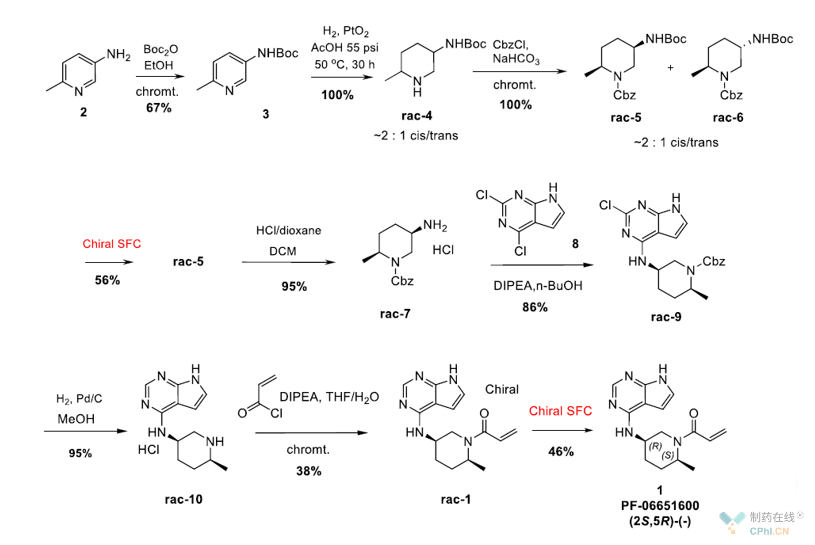
图一 PF-06651600初始合成路线
工艺路线的优化策略
引入具有迈克尔加成受体官能团(如丙烯酰胺官能团)的API是一种常见的药物设计策略。但是这类API化合物面临的挑战在开发的早期就显而易见,其在制备,纯化,储存中可观察到由于迈克尔加成而产生的杂质。由于化合物1以无定形游离碱的形式存在,这加剧了迈克尔加成杂质的生成。
在最后一步API合成步骤中,研究人员分离鉴别了三种主要杂质,其中11和12是与工艺相关的杂质,可以通过纯化1除去。然而,迈克尔加成杂质13却可随着时间的推移而有所增加。
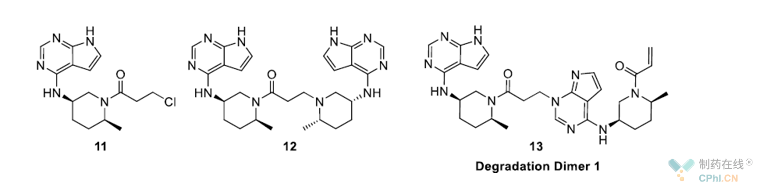
图二 API合成步骤中的主要杂质
为了在改善路线经济性和提高API交付速度之间取得平衡,研究人员认为需要解决早期开发阶段出现的以下问题:
(1)寻找经济有效的氢化条件以还原吡啶中间体,同时改善顺/反立体选择性;
(2)确定可扩大化生产的经典拆分方法,以在合成路线早期分离富对映体(enantioenriched)的哌 啶,从而避免手性色谱分离;
(3)提高丙烯酰胺合成步骤收率;
(4)规避所有的色谱纯化步骤;
(5)确定1的稳定晶型并且开发可靠的结晶过程对其进行分离。
PF-06651600工艺路线优化
经典拆分方法制备中间体7
为提高中间体3的收率,研究人员优化了Boc酸酐投料量以及反应温度。另外,将反应溶剂从乙醇变成THF与氯化铵水溶液(含有6当量的水和0.03当量的NH4Cl)的混合体系,可以抑制脲基副产物16的形成。通过庚烷/乙酸乙酯体系中重结晶,以95%分离产率得到中间体3,纯度> 99.5%。
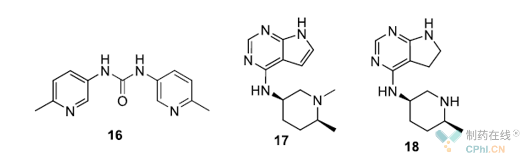
图三 工艺路线中确证的杂质
目前,使用均相Rh或Ir催化剂通过不对称氢化将吡啶对映选择性还原为哌 啶的实例不多,而以2,5-二取代吡啶作为底物使不对称氢化更具挑战性。在经初步的评估之后,研究人员将重点转向在外消旋氢化条件下替换昂贵的PtO2催化剂,并尽可能地提高顺式/反式非对映异构体的比例。最终,选择使用5%Rh / C作为催化剂,在50℃,50 psi氢气压力下,于乙醇/乙酸为9:1的混合溶剂体系中进行该反应。该条件不仅使顺/反比提高到2.8:1,而且在大规模生产中可重现性更好。使用该反应条件,研究人员在10h内成功完成了对>100Kg 中间体3的氢化。在反应完成后,通过与正庚烷共沸蒸馏将乙酸部分除去,得到粗品rac-4无色溶液(乙醇/乙酸=20:1),直接用于下一步拆分。
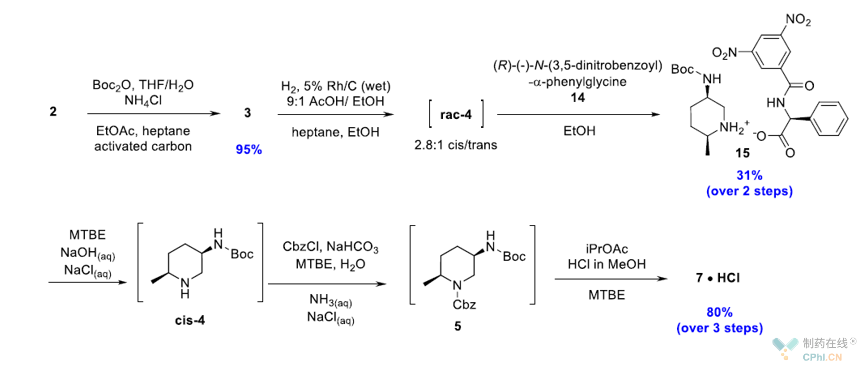
图四 中间体7的合成路线
研究人员共筛选了91种手性酸,最终选择以(R)-2-(3,5-二硝基苯甲酰胺基)-2-苯基乙酸14作为拆分剂。进一步的研究发现,采用高速搅拌和添加高纯度中间体15作为晶种的方法可以提高中间体15的光学纯度。当结晶的浆液在70℃下搅拌时间由3h增加到16 h时,观察到产品ee值从 98%下降到91%。这表明结晶过程存在一个动力学过程,其对映选择性高于热力学平衡所能提供的对映选择性。因此,为了保证重现性,研究人员开发了以下流程:首先向70°C 的14(0.45当量)乙醇溶液中加入少量中间体15晶种,然后在高速搅拌下将rac-4溶液缓慢滴加至该体系中,得到15的白色浆液。而后将白色浆液在70°C下搅拌不超过3.5小时,并在5小时内线性冷却至22°C。离心得到15白色固体,ee为97.1%。 将得到的15在热乙醇中进行打浆纯化,最终得到15的白色结晶固体,ee值99.1%。该流程在>50 kg规模多次重复验证。从3得到15的总产率为31%。
由中间体15到7·HCl的三步转化通过叠缩工艺(telescoped process)完成。首先用MTBE与 NaOH水溶液中和15,得到顺式-4游离碱的MTBE溶液。而后得到Cbz保护的中间体5。将溶剂从MTBE替换为乙酸异丙酯,通过HCl的甲醇溶液除去N-Boc基团,得到7·HCl,并进一步在 MTBE体系中结晶析出。该流程能够以80%的产率获得白色粉末状的7·HCl,同时提高产品的顺式/反式比例(100%顺式)和光学纯度(99.8%ee)。
中间体10的合成
中间体7与8在水和MIBK双相体系(5:1(v / v))中进行亲和反应,以K2CO3(3.2当量)作为碱合成中间体9。该反应在90℃下进行22小时。 反应完成后,用乙酸乙酯萃取产物9,共沸除去大部分MIBK后,在甲醇和水的混合溶剂中结晶。进一步研究表明,残留的甲醇可在随后的氢解步骤中生成N-甲基化杂质17。该杂质是通过甲醇脱水为甲醛,甲醛与N反应生成亚胺后还原而形成的。在氢不饱和的条件下,如在没有清除二氧化碳副产物或高催化剂负载下(支持脱水成甲醛),会增加17的生成。但是氢气本身也促进17的产生( 还原胺化的机理)。为避免形成17,研究人员将中间体9在水中进行打浆,然后干燥,以确保除去残留的甲醇。该步骤产率约为89%,纯度大于99.5%,可实现最大规模100Kg产品的制备。
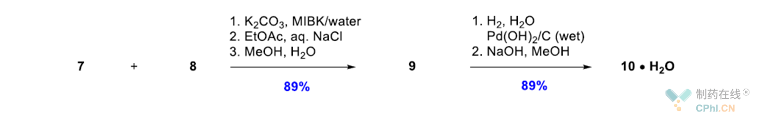
图五 中间体10的合成路线
在原合成工艺中,使用干燥的Pd/C催化剂在甲醇/THF体系中进行的氢解反应经常不能使使氯化物完全氢解,因此常需要将反应物过滤后再用催化剂进行第二轮氢解,以实现完全转化。 经筛选发现,使用10%的Pd(OH)2/C湿催化剂,在丁醇和水体系下能够更高效的完成氢化。反应完成后,过滤反应液除掉催化剂,然后用甲醇稀释。用氢氧化钠中和体系中的盐酸,并使产物结晶为一水合物(10·H2O)。在50kg条件下,10·H2O的收率为89%,纯度为99.7%。
API的合成
对于API的合成步骤,研究人员工作重点是确定更好的酰化试剂,碱和有机溶剂,同时尽量在Schotten?Baumann条件下进行反应。研究者选择已商业化的丙烯酰氯和3-氯丙酰氯作为酰化剂对反应条件进行筛选,二者最优反应条件分别列于表一中。通过比较这两种方法发现,当中间体10与3-氯丙酰氯酰化形成中间体11时,产物1的浓度最低,因此可避免产物1与10反应生成杂质12。由于11到1的部分转化,最终反应混合物中仅观察到1%副产物12。与此相反,丙烯酰氯直接酰化生成1的反应体系生成了10%的副产物12,这可能是因为产物1的浓度太高导致其与10的反应速度变快。类似地,在B条件下,杂质13的初始含量(形成速度比12慢)也降低了约90%。

表一 酰基化试剂的筛选
而后,在条件B的反应混合物中加入2 N NaOH(3.05当量)水溶液,20°C搅拌20h,化合物 11能够顺利地转化为产物1,同时其他杂质(包括13)生成较少。反应终点化合物11:12:1:13的分布为0.2%:1.1%:93.5%:0.2%。另外,AMES测试表明,中间体11不是遗传**杂质,可被视为正常的工艺杂质。综上所述,条件B被选择作为放大生产的工艺路线,并在50 kg规模上证明了其可重复性。
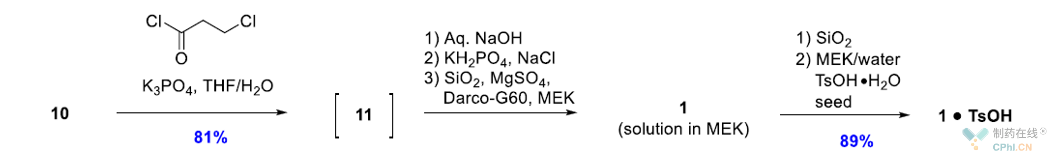
图六 API合成步骤
在得到中间体11后,研究人员开发了一种带有部分纯化功能的后处理方式,以提供1的游离碱。在消除反应完成后,首先加入KH2PO4 (5.3 equiv)调节pH至6.5,盐水洗涤,有机相纯度提高到96.2%。将溶剂从THF切换到MEK后,与硅胶(130 wt %)、MgSO4 (30 wt %)、活性炭Darco G-60 (15 wt %)搅拌,进一步纯化,过滤。滤液浓缩后,得到1在MEK中的质量分数为30 wt %的溶液,纯度为98.5%,产率为81%。由于1不稳定,且在高温下易生成杂质13,因此路线中所有蒸馏操作都是在低温(<30℃)下进行并尽量缩短1的储存时间。
为了使API能够更加稳定,研究人员进一步筛选了众多化合物1的盐,并最终确定以对甲苯磺酸与1成盐。最初的3Kg规模放大使用了1的30 wt%的MEK溶液,该溶液已在10°C下保存了3周,纯度从98.5%降至98.0%,二聚体杂质13从0.2%升高至0.5%。在成盐之前,首先用SiO2(25 wt%)对该溶液进行预处理以除去部分杂质13(0.5%至0.2%)和其他少量未鉴定的杂质。而后,将MEK和水添加到此预处理后的1的MEK溶液中,并滴加TsOH·H2O(1.1当量)的MEK溶液。此时,水和MEK的溶剂组成为5.3%∶94.7%(w/w)。加入晶种(4wt%)后,将混合物高速搅拌以形成分散良好的白色浆液。缓慢加入MEK使溶剂组成达到2.3%∶97.7%(w / w)。过滤该混合溶液得到白色固体,产率为89%,纯度约99.6%。由化合物10到成盐的两步反应经多批> 50 kg制备得到了进一步验证。该工艺路线制备的PF-06651600甲苯磺酸盐为API储存和片剂制剂均提供了令人满意的稳定性。
结语
通过工艺的持续改进,路线总收率从5%提高到14%,并且已经生产了多批次公斤级的API产品,为正在进行的临床研究提供了有力的支撑。
参考文献
1. ACS Chemical Biology, 2019, 14(6), 1235-1242.
2. Org. Process Res. Dev., 2019, 23, 1872-1880.
点击下图进行CPhI & P-MEC China 2020观众预登记!立省100元门票




合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























