Alectinib简介
Alectinib是罗氏公司控股的日本中外制药Chugai公司研发的口服ALK抑制剂。Alectinib分子结构中包含一个独特的苯并咔唑衍生物的骨架结构,这种结构能够与 ALK 激酶区完全结合,从而表现出对 ALK 更高的选择性及抑制效果。
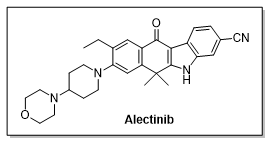
2015年12月,Alectinib被美国FDA批准上市,主要用于克唑替尼耐药后的非小细胞肺癌(NSCLC)患者的治疗。近年来,随着临床研究的不断深,Alectinib又于2017年11月被批准用于晚期ALK突变的非小细胞肺癌一线治疗。在国内,Alectinib于2018年8月由中国CFDA批准上市,用于晚期ALK突变的非小细胞肺癌一线治疗。
既往研究结果显示,Alectinib不仅能显著延长NSCLC患者的无进展生存期,而且在已经发生脑转的患者中,Alectinib一线使用后的疗效也十分显著。和同类的其他ALK抑制剂,例如克唑替尼、塞瑞替尼、布加替尼相比,Alectinib的无进展生存期最长,治疗效果最 佳,备受临床关注。
与此同时,Alectinib具有高效低毒的特点,中位PFS可达到34.8个月,是目前ALK+ NSCLC患者的一线优选方案,也是克唑替尼耐药后的优选药物。在不良反应方面,Alectinib常见的不良反应主要有便秘、疲乏、水肿等,主要为1-2级不良反应。
2020年,Alectinib已全线降价并进入国家乙类医保,这在很大程度上减轻了广大患者的经济负担。
Alectinib合成路线一览
Bioorganic and Medicinal Chemistry ,2012 ,20 ,1271-1280
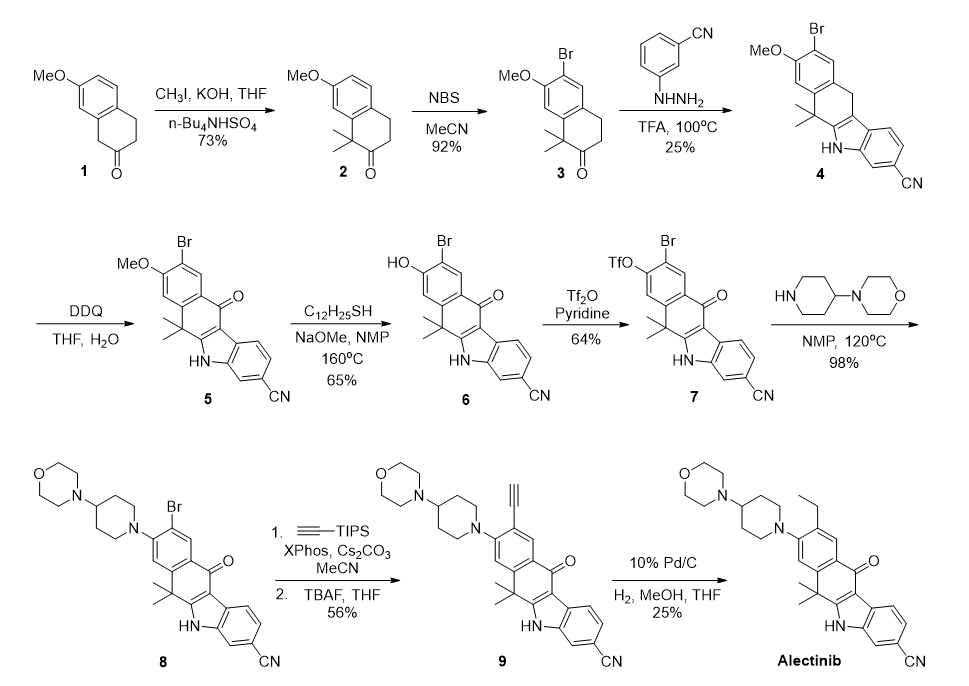
图一 实验室发现路线
该路线为药物化学发现阶段实验室合成路线,以7-甲氧基-2-四氢萘酮1为起始化合物,经9步线性步骤,得到Alectinib的游离碱产物,总收率约为1%。该路线总收率过低,在中间体3与肼基苯发生Fischer吲哚反应的步骤中,由于区域选择性较低,得到一对区域异构体,经硅胶柱层析纯化后得到目标产物4,收率只有25%。此外,中间体8与TIPS-乙炔经Sonigashira反应,TBAF脱硅基反应生成乙炔中间体9,然后将乙炔中间体9加氢还原从而引入乙基,该三步收率为14%,引入乙基的效率太低。
EP 3556754 A1
为进一步优化乙基引入方法,费森尤斯卡比公司报道了通过"一锅法"由中间体6合成Alectinib的工艺路线。
在干燥的Schlenk管中,在氮气下依次加入中间体6、PdCl2(PPh3)、三甲基甲硅烷基乙炔、TEA和DMF。封闭Schlenk管,并转移到80℃的油浴中。搅拌反应直至反应物完全转化。将管从油浴中移出并在室温下冷却。在氮气下,添加MeOH和K2CO3,并将反应搅拌2小时。将溶液转移至高压釜中,并将THF和Pd/C 10%加入反应混合物中。在2.5 bar的压力下将高压釜充满H2并将反应搅拌8小时。从反应器中释放出氢气,并将反应混合物过滤到硅藻土垫上,并转移到烧瓶中。后处理,从热MeOH中重结晶出固体,获得纯产物,收率为69%。
专利中该路线仅能够得到100mg左右产品,不确定是否适用于更大规模的生产。
U.S 9126931 B2
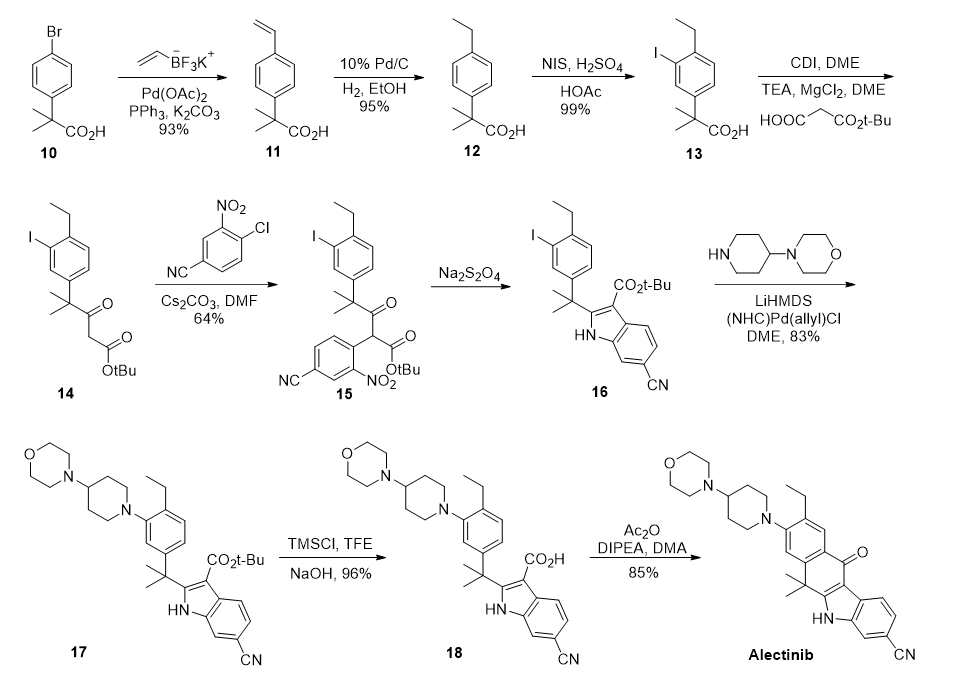
图二 U.S 9126931 B2 路线
基于实验室路线的缺点,U.S 9126931 B2中随后报道了一条改进路线,对乙基引入以及吲哚杂环的构建进行了优化。
该路线以溴 基苯 乙 酸化合物10作为起始原料,利用Molander variation of Suzuki-Miyaura 交叉偶联反应,与乙烯基三氟硼酸偶联得到烯基中间体11,随后经还原氢化得到乙基中间体12。两步收率均大于90%。得到中间体12后,通过碘取代,碳链延伸,亲核取代,硝基还原然后关环四个步骤,构建吲哚杂环中间体16。
而后,通过Pd催化的C-N交叉偶联将哌 啶基侧链与16连接,得到中间体17。以三氟乙醇为溶剂,TMSCl脱保护得到羧酸中间体18。最终,通过醋 酸 酐介导的分子内Friedel-Crafts反应闭环得到目标产物alectinib。
根据专利报道,该路线每批投料量最高可达1.4Kg,路线总收率为38%,平均每步收率89%。
WO 2016074532
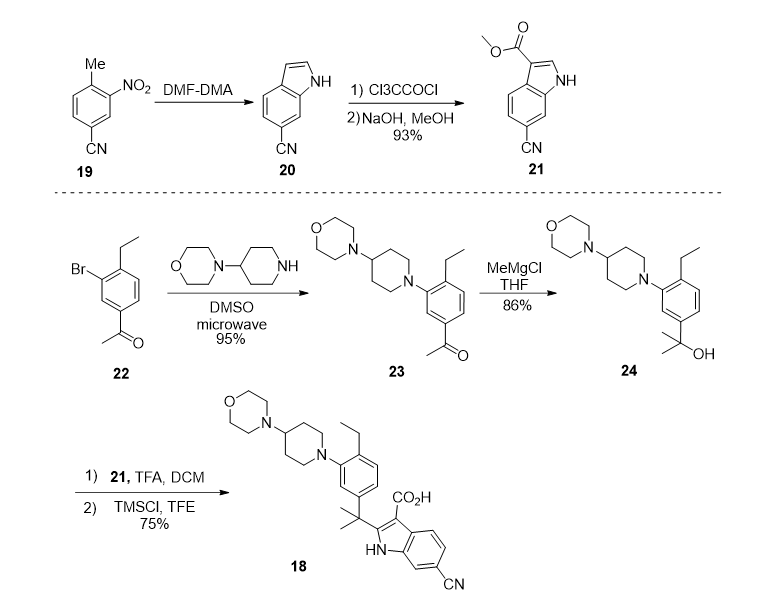
图三 WO 2016074532 路线
专利WO 2016074532对上述路线中关键中间体18的合成方法给出了另一条合成路线。该合成路线以硝基苯基化合物19作为起始原料,经Leimgruber-Batcho反应,构建吲哚杂环20。而后经酰基化反应得到酯基中间体21。
与此同时,作者以化合物22作为起始原料,与哌 啶杂环在微波条件下亲核反应得到酮基中间体23。而后在甲基格氏试剂的作用下,生成羟基化合物24。最终,中间体24与吲哚中间体21经酸催化的傅克反应,生成中间体18。
该路线需要用到微波催化反应,在工业化生产中较难实现。起始原料22较为昂贵,比较难从商业化途径获得,这都限制了该路线的应用价值。
CN106518842A
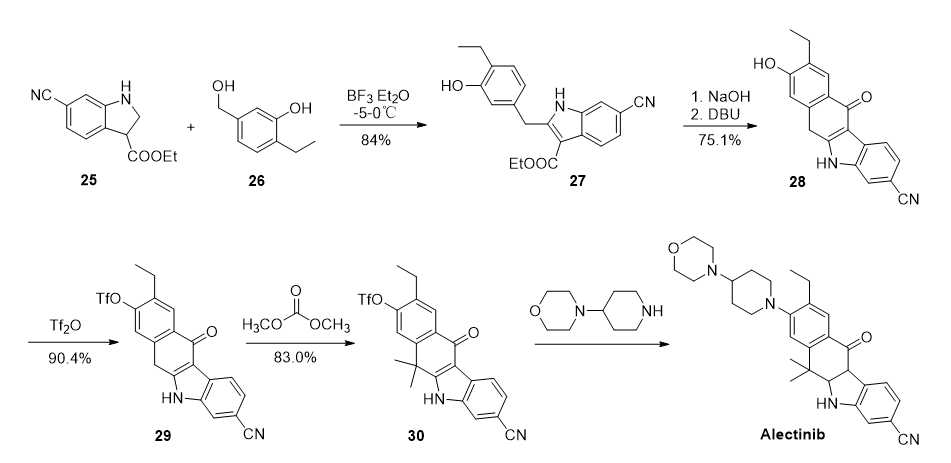
图四 CN106518842路线
中国专利CN106518842以6-氰基-1H-吲哚-3-甲酸乙酯25为原料,与4-乙基-3-羟基苄醇26经过缩合得到吲哚-3-甲酸乙酯中间体27,然后经酯基水解、酸化成酸、DBU作用下环化得到28,再经三氟乙酸酐保护羟基、碳酸二甲酯甲基化、4-(4-哌 啶基)吗啉取代反应制得Alectinib,总收率为38.8%。
参考文献
1. Org. Process Res. Dev. 2016, 20, 11, 1855-1869
2. Bioorganic and Medicinal Chemistry ,2012 ,20 ,1271-1280
3. EP 3556754 A1
4. U.S 9126931 B2
5. WO 2016074532
6. CN 106518842 A



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























