PART. 01
背景
口服固体制剂通常可分为常释制剂(Immediate Release, IR)和调释制剂(Modified Release, MR)。常释(IR)制剂的设计目的是使药物在胃肠道中自由溶解,而不是有意延缓或延长给药后药物的溶出和吸收。常释制剂可能是快速溶出(rapid dissolution)和非常快速溶出(very rapid dissolution)的,也可能是缓慢溶出的。对于快速溶解的IR制剂,ICHQ6A定义为在pH1.2、4.8、6.8的介质中,15min内溶出量均不低于标示量的80%(FDA GUIDANCE中的定义略有偏差,见附录)。调释制剂则包含更大范围的口服药物制剂,如肠溶衣片、缓释制剂、控释制剂等。
固体制剂要发挥药效必须先在胃肠道溶出,然后药物分子经胃肠粘膜吸收入血,经循环系统到达靶点位置发挥作用,如图1所示。药物的溶出度可能会影响生物利用度,进而影响药效。因此,溶出试验对于口服固体制剂的开发至关重要,其贯穿药品开发的整个过程。目前,只有少数口服固体制剂可能以崩解时限来检测产品的体外表现更为合适,其余产品均需要进行溶出试验。
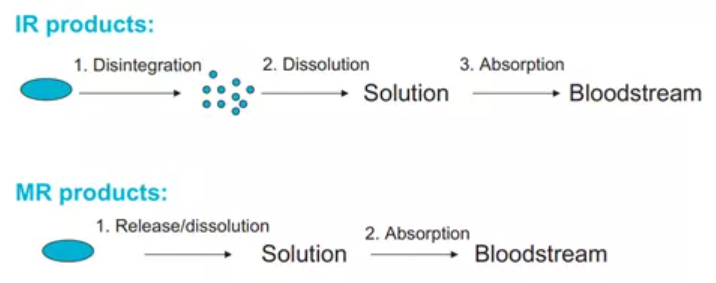
图1:固体口服制剂吸收步骤示意图
PART. 02
溶出试验的目的
溶出试验是用来比较口服固体制剂体外表现的工具。进行溶出试验的目的之一是辅助设计和筛选处方。在制剂研发的前期,当片剂或胶囊的溶出试验始终达不到标准,可能指示着需要调整处方和工艺,或者溶出方法。影响溶出的因素很多,如粉末的组成、原辅料的物理性质(原辅料颗粒的粒径分布、颗粒的密度、聚合物的粘度等级、辅料的来源等)、制造工艺(湿法制粒/干法制粒/直压、制粒参数、润滑时间、压片压力、包衣参数等)、溶出方法的选择、溶出试验时的外界环境等。
我们应通过溶出实验理解药物释放机制和辨识制剂行为,除了对溶出结果进行含量分析以外,要注意实验现象的观察,如制剂或粉末漂浮、崩解的改变和锥形的堆积等。这些简单的现象可能具有深远的意义。
进行溶出试验的另一个目的是可以进行不同批次产品之间的质量控制(Quality Control)。当获得足够数量的具有不同溶出曲线的不同批次产品的药动学(PK)数据时,便有可能通过合理的溶出度试验建立起体内体外相关性(IVIVC)。在建立好IVIVC之后,体外溶出的结果可以作为产品体内表现的替代(surrogate)。在清晰的IVIVC基础上,溶出结果可以为某些BCSⅠ类和Ⅲ类快速溶出产品的注册申报以及放大生产和批准后的重大变更提供生物等效性(BE)试验豁免的依据。
这样的溶出试验不仅能减少监管部门的审评时间,还能减少厂商在BE试验上消耗的时间和金钱。但是,需要注意的是IVIVC的建立是非常困难的,建立IVIVC需要系统地了解药物分子的理化性质、生物药剂学性质和制剂处方设计及其与胃肠道的相互作用,尽管有一些成功的案例,但是由于体内溶出和吸收的复杂性,仍然有许多的产品不能建立体内体外相关性。
在没有建立准确的IVIVC的情况下,用溶出测试来作为质控手段需考虑以下几点:
1、溶出数据是方法依赖性的,对于同一个产品,不同的溶出方法可能得到具有显著差异的溶出数据。因此溶出方法的开发非常重要。
2、一般来说,通常会基于BE/BA批次设定溶出标准(specifications)。垂直比较同一产品上市后不同批次的溶出数据与之前设定的溶出标准之间的差异来进行质量控制是有意义的。通常不会去横向比较不同产品的溶出(仿制药生物等效性豁免biowaiver除外)。
3、溶出方法应具有合适的区分力,应能够检测到可能引起生物不等效的重大变化,同时应容忍常规生产的正常变化,防止不必要的产品报废。
PART. 03
溶出方法的开发
固体口服制剂最常用的方法是篮法(Apparatus Ⅰ)和桨法(Apparatus Ⅱ),如图2所示。篮法常用转速为100rpm,桨法常用转速为50-75rpm。对于会漂浮的固体制剂,如胶囊剂,使用篮法可能更为合适,若要使用桨法,应使用沉降装置抑制漂浮。对于片剂,桨法(Apparatus Ⅱ)可能使用得更多。需要注意的是,对于桨法来说,溶出杯中流体动力学性质是高度异质(heterogeneous)的,药物在杯底的位置可能改变溶出速率。如果片子偏离溶出杯中心可能导致流体动力学性质的改变,主要影响崩解过程的药物释放。
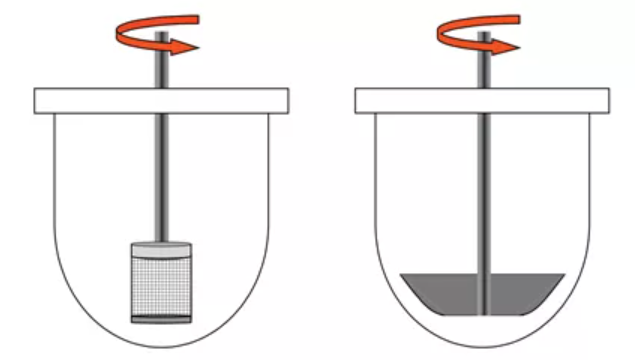
图2:篮法和桨法装置示意图
在选择合适的溶出介质之前,需要测定药物活性成分的物理和化学稳定性。决定溶出介质的组分之前,需要充分评估缓冲液、pH的影响。如需加入表面活性剂,也需研究不同表面活性剂对药物溶解度和稳定性的影响。
溶出介质的选择需满足漏槽条件,以保证溶出时有足够大的浓度梯度,更准确地反映制剂本身的性质。在此之前,需测定药物在不同pH介质中的饱和溶解度,溶解度是药物最重要的理化性质之一,药物溶解度的测定及改善,是制剂研发过程中必须做的一项工作。大家通常认为溶解度测定是一项微不足道的工作,但是要准确测定却是非常具有挑战性的,很多因素均会影响溶解度的测定,导致结果出现偏差。
测量平衡溶解度介质包括pH范围为1.2-7.4的缓冲液、模拟胃液和肠液以及纯化水。在测定饱和溶解度的过程中,需监测上清液在试验过程中的pH变化,如有需要需回调pH。如有需要,还要研究不同表面活性剂(种类和浓度)对药物溶解度的影响、评估离子效应对溶解度的影响、研究药物在其他温度下,如25℃时的溶解度。
溶解度测定的注意事项:
●溶质是否降解;
●溶质的固体形态(晶型);
●检测和矫正溶液的pH(pH-溶解度);
●选择的电解质最好不含有溶质中存在的离子,以避免同离子效应;
●缓冲液与电解质本身可能与电离的药物形成溶解度较低的盐,试验结束后确定剩余固体是否含有缓冲液与电解质的成分,评估盐型是否改变。
进行溶解度实验时,要注意观察实验现象,尤其是存在溶解又析出的情况时,转晶或转盐的风险则较大,需特别关注。
PART. 04
溶出方法开发报告
为了支持所选的溶出方法,需要在注册申报的资料中提供溶出方法开发报告,开发报告需提供以下内容:
1、生理pH范围内,API的溶解度数据。因为溶解度数据是选择合适的溶出介质以及决定是否需要表面活性剂的基础。
2、设备和方法的选择、溶出介质、介质pH、介质体积、搅拌速度、含量测定、漏槽条件、沉降装置的选择(如有需要)、酶的选择(如果需要),等等。如果加入了表面活性剂,需要提供所选择的表活种类和量的数据支持。
3、取样时间点:一般来说,对于速释制剂,选择10、15、20、30、45、60、90min;对于缓释制剂,选择1、2、4、8、12、24h。
4、证明所选溶出方法的区分能力:可以通过有意地调整生产变量(如对关键物料属性(CMAs)和关键工艺参数(CPPs)上下调整10-20%)来生产不同质量的产品,再去比较其溶出来证明溶出方法的区分力。下图显示了调整片剂硬度和粘合剂粘度对溶出度的影响。
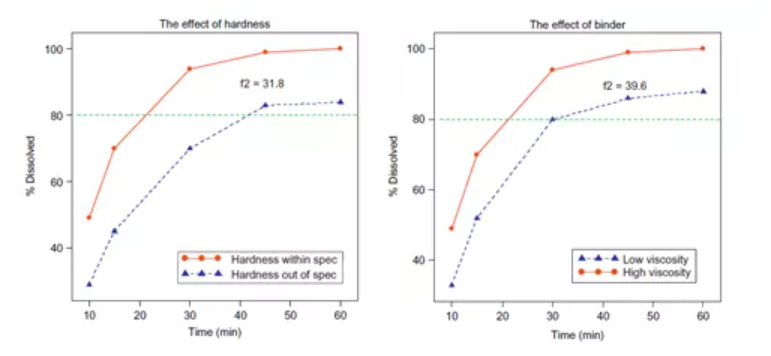
图3:片剂硬度及粘合剂粘度对于溶出度的影响
有时也会出现溶出方法过度区分的情况,如图4所示,虽然工艺变化后生产的样品与参比样品的体外溶出度存在显著差异,但其体内生物利用度却基本一致。
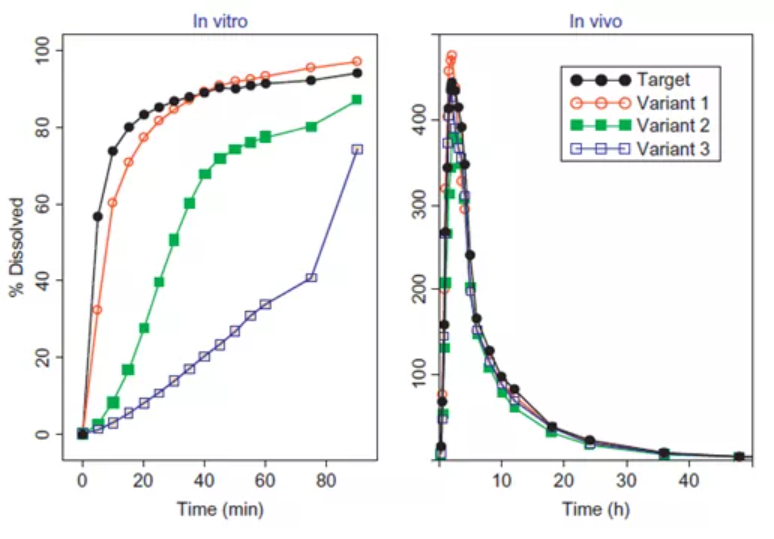
图4:过度区分的溶出方法
当然,在实际的产品开发过程中,可能会遇到速释制剂溶出过快,始终无法找到有区分力的溶出方法的情况。此时可根据ICH Q6A决策树#7执行,只要溶出度并不明显影响生物利用度,且处方和工艺的变更不影响溶出度,即可不必过多考虑溶出方法的区分力。不过此时需要收集产品在多个溶出介质中的溶出数据(如pH1.2, pH4.5, pH6.8),防止在申报过程中出现问题。
5、提供溶出方法的其他验证数据,如系统适用性、方法耐用性、介质是否需脱气、介质pH耐用性、仪器温度耐用性、滤膜吸附试验、溶液稳定性研究、重复性试验、中间精密度研究、自动和手动取样验证以及含量分析方法的验证(专属性、线性和范围、回收率等)。
PART. 05
溶出方法质控时间点
和可接受标准的选择
当设定溶出质控标准时,产品的临床表现是首先也是最重要的考虑因素。因此,应结合关键临床批的体内数据和体外溶出数据来制定溶出标准(时间点和溶出限度)。
两个常释(IR)制剂的例子:
案例1:如图5所示,与另外两批相比,临床批的溶出度最高,临床批与测试批次2的溶出曲线相似(f2因子=56.3),临床批与测试批次1的溶出曲线则不相似(f2因子=44.7)。而临床BE试验表明,与临床批相比,测试批次1的BE刚好符合BE接受标准。因此,在制定溶出标准时,可以考虑将批次1的溶出作为下限。需要注意的是,f2因子的参考最好建立在体内数据的基础上,因为两批生物不等效的产品其体外数据会有多相似,两批生物等效的产品其体外数据又会有多不相似,我们不得而知。
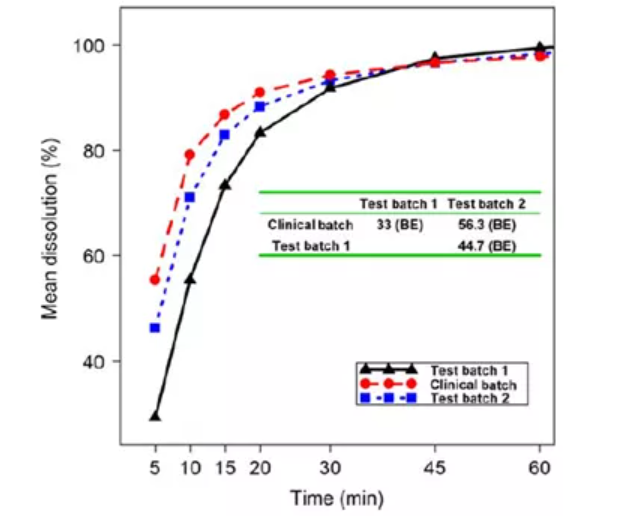
图5:三批生物利用度相同的产品的溶出曲线
案例2:如图6所示,已知批次A属于关键临床批次,已被证实安全有效,批次B与批次A生物不等效。那么在制定溶出标准时,以80%为溶出限度,便不能选取30min作为质控点,这样会导致两批产品同时被接受,而选择15min作为质控点就能拒绝批次B。
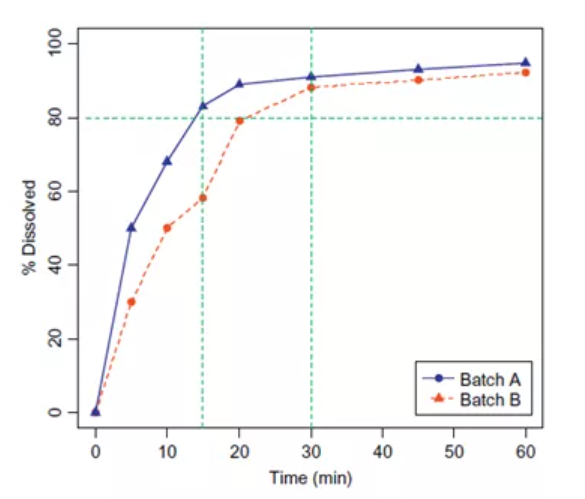
图6:生物不等效的两个批次产品的溶出
同时ICH Q6A指出,如果与崩解测试相比溶出不能作为产品质量控制的敏感指示标志,可能就需要将崩解作为质控标准。
综上所述,溶解度测定和溶出方法的开发是非常细致的工作,溶解度测定为溶出方法的选择奠定基础,而溶出实验贯穿产品开发的始终,实验必须在法规的指导下进行,才能得到准确的实验数据。
附录:
快速溶出(rapidly dissolving):用USP装置Ⅰ或Ⅱ的方法,在<900ml的水性介质中,30min内应有超过85%的药物溶解;而非常快速溶出(very rapidly dissolving)则是15min内有超过85%的药物溶解。
参考文献:
1.《普通口服固体制剂溶出度试验指导原则》
2. Yihong Qiu et al. Developing Solid Oral Dosage Forms_ PharmaceuticalTheory and Practice
3. Pharmaceutical Research, Vol. 19, No. 7, July 2002
4.Waiver-of-In-Vivo-Bioavailability-and-Bioequivalence-Studies-for-Immediate-Release-Solid-Oral-Dosage-Forms-Based-on-a-Biopharmaceutics-Classification-System.-Guidance-for-Industry
5. Q6A:质量标准:新原料药和新药制剂的检测方法和可接受标准:化学药物



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























