近些年,随着国家政策的一系列调整,创新药的开发得到了前所未有的提速,每年IND新增品种数量更是成倍申报。新药开发的面纱,逐渐被行业摘去,但透露出来的往往集中于药学,即CMC部分,而临床前&临床相关工作的组织和背后逻辑,依然相对神秘。在此,笔者根据即往经验及对相关法规&指导原则的理解,与大家共同分享,创新药首次申请临床,申办方需要提供的临床专业内容。
01
CTD资料需要哪些内容?
CTD格式文件模块共5个,分别为模块1~行政管理信息、模块2~通用技术文档总结、模块3~质量研究信息、模块4~非临床试验报告、模块5~临床研究报告;其中,对于新药的首次临床申请,与临床内容最为直接相关的部分为模块2的2.5临床综述、2.7临床总结,以及模块5的临床研究报告。
对于CTD通用技术文档的组织,在临床综述、临床总结、临床报告的基础上,内容可具体到临床试验计划中的开发计划(可包括I~III期)、开发路径、开发时间;临床研究方案中的剂量选择依据(包括起始剂量、最大耐受剂量、分组、剂量递增)、盲法(包括揭盲、评价指标、受试者选择)、安全/耐受性评价、数据分析、不良事件、伦理,等;以及非常重要的研究者手册。同时,还要对同类药物临床研究结果(包括I期/II期/III期)、临床安全性进行重点信息汇总。
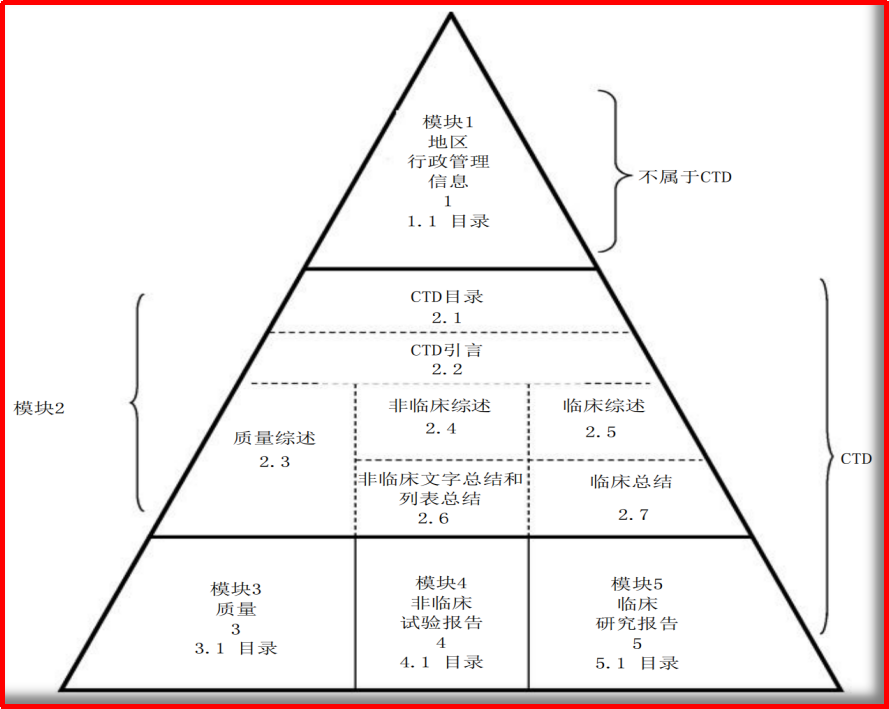
图1.1
ICH-CTD通用技术文档的组织图示
02
M4E指导原则关于新药临床I期的要求
模块2中的临床综述和临床总结,是申报资料临床内容的位置,其涵盖的内容不只是针对新药的临床I期,所以首次临床申请可根据M4E中的内容进行客观描述,即有余地有选择的撰写。
临床综述,应:
1)描述和解释药物临床开发的总体思路,包括关键临床研究设计决策;
2)评价研究设计与实施的质量,包括执行药物临床试验质量管理规范(GCP)情况的声明;
3)简述临床发现,包括重要的局限性(例如缺少与特别相关的阳性对照药的比较,或者缺少某些患者人群、相关终点或联合治疗的信息);
4)依据相关临床研究的结论提供获益与风险评估,包括解释有效性和安全性结果如何支持拟定剂量和目标适应症,以及评价如何利用说明书和其他方法优化获益和管理风险;
5)提出开发中遇到的特殊有效性或安全性问题,并说明这些问题是如何评价和解决的;
6)探讨未解决的问题,说明其不影响批准的理由,并提供解决这些问题的计划;
7)解释说明书中重要或不常见部分的依据。
临床总结,是CTD中对所有临床信息的详实总结。这包括ICH E3临床研究报告中提供的信息;从任何荟萃分析或其他交叉研究分析获得的信息,其完整的报告已经包含在模块5中;以及在其他国家或地区的上市后数据。本文件在进行研究结果间的比较和分析时,应重点关注实际观察到的数据。相比之下,CTD临床综述文件应提供临床研究项目及其结果的关键分析,包括对临床发现的讨论和解读,以及试验药物的临床定位。

图2.1
ICH-CTD模块2关于临床的资料内容
模块5中的临床研究报告和相关信息的组织,是推荐的一个特定构架用于组织临床研究报告和相关信息,可以简化申报资料的准备和审评,并保证资料的完整性;报告的布局应由研究主要目的决定;每项研究报告应仅在一个章节中描述;如果某项研究有多个研究目的,应在多个章节中交叉参考这项研究;当没有适于某个章节或分章节的研究报告或信息时,应提供“不适用”或“未进行研究”等解释。

图2.2
ICH-CTD模块5关于临床的资料内容
03
国内法规&指南相关重要文件
对于I期临床试验,CDE曾相继发布《药物I期临床试验管理指导原则(试行)》、《新药I期临床试验申请技术指导(草案)》、《抗肿瘤新药首次人体临床试验申请临床相关资料准备建议》,等;这里着重介绍下后二者。
➣ 《新药I期临床试验申请技术指导》
2016年9月,国家食品药品监督管理总局(CFDA)发布《新药I期临床试验申请技术指导(草案)》,其涵盖了IND提交所需的所有具体信息,信息量很大,而与临床相关的内容,主要有“介绍性说明和总体研究计划、研究者手册、方案、研究药物既往在人体使用的经验、临床试验暂停要求、伦理要求、IND安全性报告、IND年度报告、撤回终止暂停或者重启IND”;同时,附件信息还有“药品注册临床试验申报资料信息表”、“研究者声明表”。
该技术指导最大的背景为“保护研究受试者的权利和安全,从而保证临床试验的质量以科学、充分地评价药物的有效性和安全性”。

图3.1
《新药I期临床试验申请技术指导(草案)》
相关临床内容
➣ 《抗肿瘤新药首次人体临床试验申请临床相关资料准备建议》
2018年2月,CDE为积极贯彻落实中办、国办联合印发《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》特组织撰写发布《抗肿瘤新药首次人体临床试验申请临床相关资料准备建议》。PS:在当时创新药临床试验申请中大约占40%。
该《建议》的总体要求为:在以方案为核心的临床审评中,建议同时提供《总体研发计划》对临床研发策略进行概要性总结,并有《临床综述资料》、《风险控制计划》、《研究者手册》、《知情同意书》和伦理委员会相关材料等对试验方案予以支持。
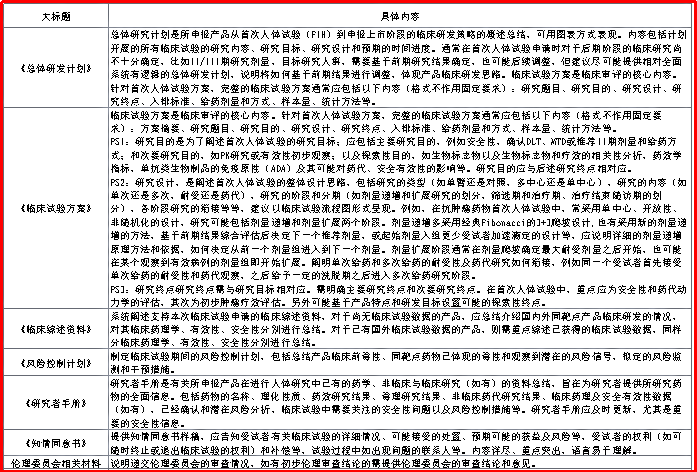
图3.2
《抗肿瘤新药首次人体临床试验申请临床相关资料准备建议》
临床相关内容
04
小结
综上,即为创新药首次临床申请所需要资料的大致方向。首先,要说明,上述条目存在一定的审查时效性,但内容和本质实际上是不变的。其次,首次临床资料的大量内容都是建立在非临床试验结果之上,故为了更好的开展临床工作,对非临床的内容和数据也要有较好的认识基础。最后,本文介绍的内容整体来说,还是以初步入门了解为主,真正想了解临床资料内容,除实打实地参与临床相关工作内容外,还需要对方案、过程、数据,进行汇总、联系、贯穿,由此才能推出一个药的有效性和安全性。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























