嵌合抗原受体T细胞((CAR-T)疗法在血液系统恶性肿瘤的治疗中取得了一定的突破,但是同期PD-1产品,仅Keytruda在2021年就实现170多亿美元的营收,但是与此相比所有上市的CAR-T产品2021年总营收也难望其项背。原因很多(治疗费用高昂、产能不足等),其中重要的一个因素是适应症局限于血液瘤,且大部分都是末线治疗。所有针对实体瘤的CAR-T还处于探索阶段,目前CAR-T治疗在实体瘤中面临着很多挑战,包括免疫抑制性肿瘤微环境(TME)、肿瘤抗原异质性、基质障碍和肿瘤可及性,以及靶向/脱靶等困境。
A 抑制性肿瘤微环境
与血液系统恶性肿瘤不同,实体瘤中癌细胞的可及性受到限制,其中还包括一些物理障碍,比如肿瘤脉管系统和细胞外基质,这些会阻碍注入的CAR-T细胞渗透到肿瘤组织。实体瘤是一种器官样、杂乱无章的结构,由增殖的肿瘤细胞组成,周围有支持性基质细胞和滋养肿瘤新血管系统的血管,肿瘤的生长可以由先天性和适应性成分控制。实体瘤中先天免疫细胞主要有:中性粒细胞、巨噬细胞、树突细胞(DC)、肥大细胞、自然杀伤细胞(NK细胞)和髓源性抑制细胞(MDSC)。适应性免疫细胞:T和B细胞以及调节性T细胞(Tregs)。所有这些免疫细胞都与构成TME的非肿瘤基质细胞相关:内皮细胞、成纤维细胞、周细胞和间充质细胞。上述这些细胞及其分泌的因子和分子构成了TME。CAR-T细胞在实体恶性肿瘤治疗中遇到的主要障碍,是TME通过抑制性可溶性因子和过度表达阴性免疫检查点来阻止T淋巴细胞向肿瘤运输和渗透建立免疫抑制环境,T细胞有效浸润到肿瘤基质中是保持被注入T细胞的抗肿瘤活性和癌症免疫治疗成功的关键步骤。
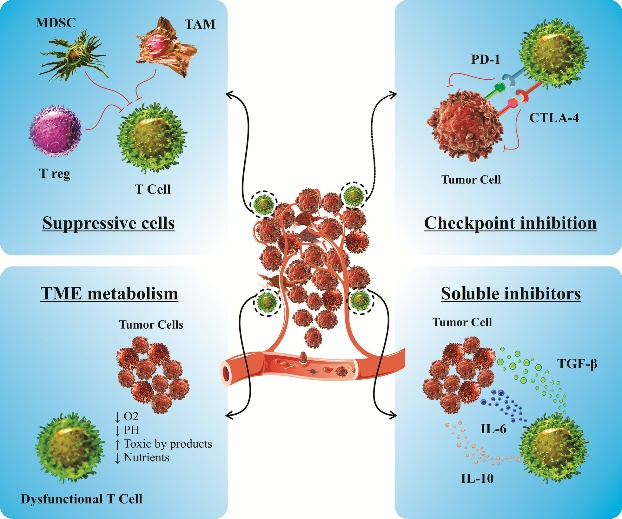
图1 肿瘤免疫抑制微环境
➣肿瘤微环境缺氧
(1)缺氧是影响肿瘤进展并改变癌症患者治疗结果的肿瘤微环境的标志。缺氧诱导因子(HIF)的稳定性和核转位,它通过与缺氧诱导基因启动子中的缺氧反应元件(HRE)区域结合来进一步调节基因转录,导致细胞适应环境变化。另一方面,感染和炎症过程中产生的T细胞受体(TCR)激活信号或细胞因子可以调节HIF的合成和稳定性,进而影响T淋巴细胞的激活和分化。在持续感染或肿瘤微环境下,TCR介导的T细胞刺激和缺氧条件增加了HIF丰度和细胞**T淋巴细胞(CTL)的效应功能。这时,Von Hippel-Lindau (VHL)因子会部分抑制T细胞反应,以保护身体免受过度反应的破坏性。T淋巴细胞中HIF1α的稳定与糖酵解酶(如作为葡萄糖转运蛋白的GLUT-1)表达的增强和氧化磷酸化速率的降低平行,因此可以通过缺氧依赖和缺氧非依赖途径增加HIF水平和活性,作为代谢途径的调节剂,并参与T细胞增殖、分化和效应活性。
(2)缺氧会促进三磷酸腺苷(ATP)分解和抑制腺苷激酶(将腺苷磷酸化位AMP),从而导致腺苷增强。肿瘤微环境中低氧来源积累的腺苷与T细胞表达的特定受体(即A2AR和A2BR)的相互作用会干扰TCR信号传导,从而抑制T细胞的抗肿瘤活性。重要的是,腺苷和G蛋白偶联受体(GPCR)的相互作用激活蛋白酶A(PKA),后者随后调节TCR下游信号传导。如果CAR-T细胞表达RIAD(调节亚基I锚定破坏肽)能阻止PKA在免疫突触中的定位,可以削弱腺苷对间皮素-CAR-T细胞活性的抑制作用;另外使用小分子如BAY 60–6583(腺苷A2B受体激动剂)可以通过影响多个靶点来提高CAR-T细胞的治疗效果。
(3)免疫检查点PDL1分子以HIF-1α依赖方式上调,缺氧的环境阻碍了浸润T细胞的功能。通过PD1阻断消除PD1与PDL1的相互作用可以增强肿瘤浸润T淋巴细胞上A2AR的表达,导致对积累的腺苷免疫抑制的敏感性增强。因此同时靶向PD1/PDL1轴和腺苷A2A(通过遗传或药理学方法),可以改善CAR-T细胞治疗中的T细胞性能。
B 限制进入肿瘤细胞
➣细胞外基质
细胞外基质(ECM)作为肿瘤周围基质的一部分,由纤维蛋白、糖蛋白、多糖和蛋白聚糖组成。恶性组织中ECM成分的表达和密度增加,特别是透明质酸和胶原蛋白的过度产生和沉积,阻碍了治疗剂的渗透。此外胶原蛋白密度升高会损害T细胞的增殖和细胞毒活性,并诱导调节表型,而且CAR-T细胞制造的策略也可能造成ECM降解酶表达下调。鉴于这些事实,应用ECM降解酶(如透明质酸酶和胶原酶)可以降低ECM硬度并促进抗癌剂的输送。Caruana及其同时在CAR修饰的T细胞中诱导乙酰肝素酶 (HPSE) 表达,从而提高了共表达 CAR T细胞(即表达的CAR和HPSE)ECM降解能力和抗肿瘤活性。
➣肿瘤血管
癌细胞的细胞分裂不受控制,需要形成新的血管来获取营养和氧气。异常脉管系统的发展,以及参与T细胞外渗的黏附分子的下调(在bFGF和VEGF等血管生成因子的影响下),成为T细胞渗透到肿瘤床的物理屏障。此外,肿瘤微环境中的内皮细胞促进FasL和抑制性分子(如PD-L1、TIM3、IDO-1、PGE2和IL-10)的表达,从而抑制效应T细胞活性。肿瘤血管的这些特征部分依赖于VEGF产生及其受体的过度表达。因此针对VEGFR1和VEGFR2 设计的CAR,通过限制营养和氧气来破坏肿瘤血管和减少肿瘤细胞增殖方面显示出不错的效果。因此,可以通过同时输注VEGFR2特异性CAR-T细胞和抗原特异性TCR转导的T 细胞,增加肿瘤特异性T细胞的浸润、持久性和抗肿瘤活性来改善肿瘤特异性免疫治疗的结果。
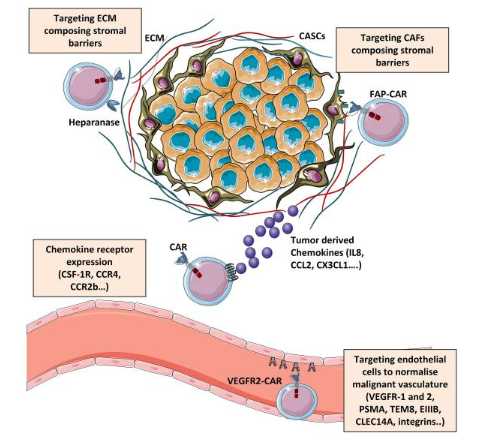
图2 增强肿瘤运输和渗透的策略
➣癌症相关成纤维细胞
癌症相关成纤维细胞(CAF)是肿瘤基质中最丰富的成分之一,代表了一种反应性的肿瘤相关成纤维细胞群,它分泌各种活性因子(VEGF和PDGF),促进肿瘤的发展、转移和治疗抵抗。CAF表达可以被免疫疗法靶向各种分子,其中FAP是最有希望的靶点。它是一种细胞表面丝氨酸蛋白酶,在人类各种癌症的相关基质细胞(CASC)上高度表达,例如:肺癌、前列腺癌、胰 腺癌、结肠直肠癌和卵巢癌。下图中描述的策略靶向CAF的关键信号和效应器,旨在抑制CAF的功能,例如细胞因子 (TGFβ) 和生长因子途径 (VEGF、PDGF)。如,可以用单克隆抗体 (MAb) 靶向CAF衍生的细胞外基质蛋白(MMP)和相关信号传导,以诱导基质耗竭和增加免疫T细胞浸润。FAP靶向旨在阻断CAF在TME中发挥肿瘤促进作用的能力,可以通过使用MAb/抗体-药物偶联物、免疫偶联物或肽-药物复合物、FAP特异性CAR-T细胞或基因敲除策略来完成。
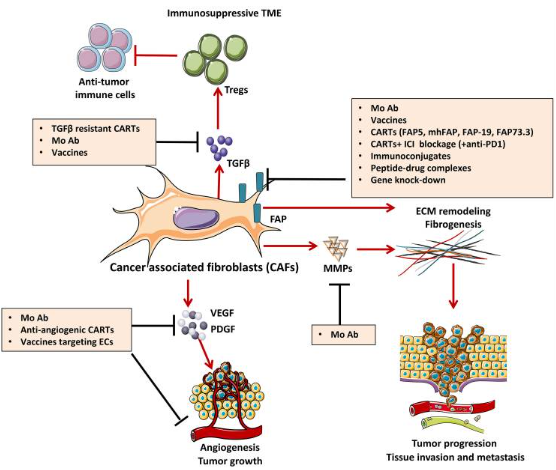
图3 抵消 CAF 致瘤效应的策略
C 存在免疫抑制细胞
➣调节性T细胞
Tregs参与肿瘤的免疫抑制环境,具有免疫抑制特征,例如分泌IL-10和TGF-β,以及竞争效应T细胞增殖所需的IL-2,抑制了注入的T细胞功能。Tregs是高度依赖IL-2的免疫抑制细胞。它们与周围环境中的IL-2结合并将其耗尽,从而通过组成表达高亲和力IL-2受体(IL2R)亚基-α(CD25)来降低T细胞的可用性。Treg细胞还产生免疫抑制细胞因子(IL-10、IL-35和TGFβ),它们可以下调T细胞和抗原呈递细胞(APC)的活性。此外,Treg细胞释放大量ATP,ATP 转化为腺苷(通过CD39和CD73),可为T细胞和APC提供免疫抑制信号。
目前来消除Treg介导的免疫抑制机制策略主要是CAR-T细胞被设计用于靶向Treg 表达的抗原将其直接消耗。其他策略基于TME的免疫调节以提高CAR-T细胞的性能:(i)CAR-T细胞表达促炎细胞因子;(ii)优化共刺激信号结构以减少IL-2分泌并削弱Treg扩增和肿瘤浸润。最后一种策略就是赋予CAR-T细胞对免疫抑制的内在抵抗力:(i)旨在破坏信号传导的显性负性受体 (DNR),或 (ii)嵌合开关受体 (CSR或Switch R) 将负信号转化为正信号,或通过使用基因组编辑工具(敲除)消除抑制性受体(如 TGFβ 受体的 PD1)的表达。
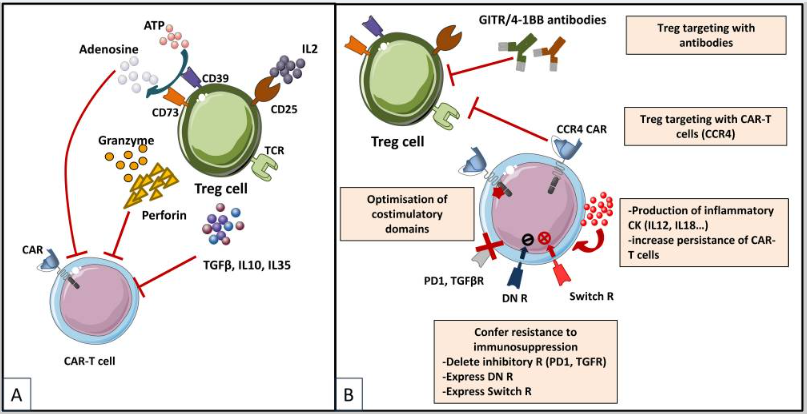
图4
Treg 在 TME 中发挥免疫抑制的机制 (A),
克服 Treg 诱导的免疫抑制的策略(B)
➣肿瘤相关巨噬细胞(TAM)
巨噬细胞是免疫系统的主要效应细胞之一,在先天性和适应性免疫反应中都发挥着关键作用,构成抵御外来病原体的第一道防线,并有助于触发适应性抗原特异性反应。巨噬细胞可以分为两个对比组:经典激活的巨噬细胞或M1巨噬细胞(促炎和抗肿瘤)和交替激活的巨噬细胞或M2巨噬细胞(抗炎和促肿瘤)。这种差异主要是由于暴漏于组织的可溶性因子或病原体衍生分子而引起极化。M1巨噬细胞是促炎细胞,通过分泌TNFα、IL-1β 和 IL-12 将细胞免疫定向到 TH1型反应在抗肿瘤免疫中发挥作用。尽管M2巨噬细胞在组织稳态中发挥作用(刺激Th2反应以消除寄生虫、免疫调节、伤口愈合和组织修复),但M2巨噬细胞也可以促进肿瘤进展。
肿瘤细胞和基质细胞可以分泌多种细胞因子和趋化因子,这些因子可将巨噬细胞募集到肿瘤细胞周围并将它们转化为TAM。当巨噬细胞被招募迁移至肿瘤间充质中后,可以通过改变自身的表型,来适应它们所处的微环境。TAM是位于TME中的M2样巨噬细胞的特殊群体,与M1和M2巨噬细胞具有一些共同的表型特征,但具有不同于这两种类型的特定转录谱。TAM通过促进遗传不稳定性和增强血管生成、纤维化、侵袭、免疫抑制和淋巴细胞排斥来增强肿瘤进展和转移。一方面,巨噬细胞产生炎性细胞因子,如IL-17和IL-23,这增加了遗传不稳定性,另一方面,它们通过分泌抑制细胞因子如TGF-β和IL-10,通过表达免疫检查点配体如PD-L1、PD-L2、B7-H4 或VISTA或通过产生活性氧(ROS),可以阻碍肿瘤免疫监视,从而阻碍T细胞介导的抗肿瘤免疫。
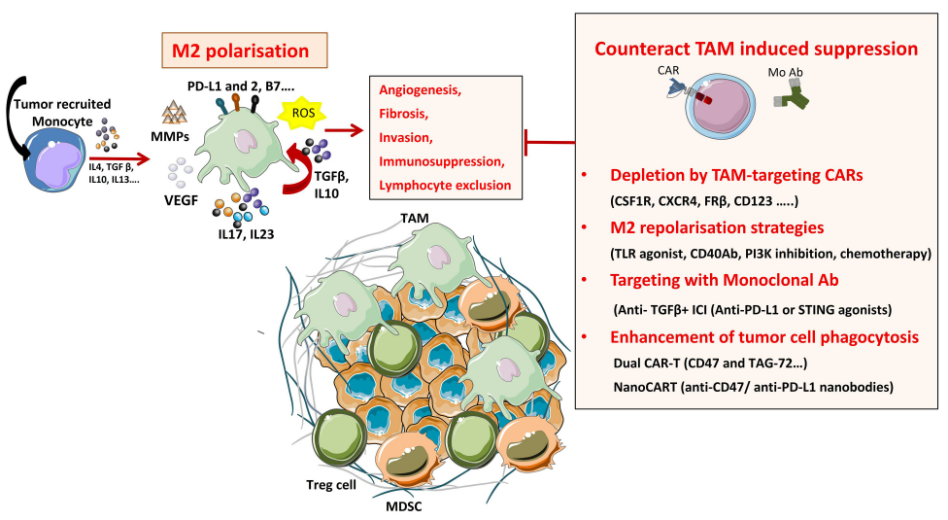
图5 克服 TAM 在 TME 中引起抑制的策略
目前针对巨噬细胞的各种治疗策略旨在耗尽或者重新极化巨噬细胞。
第一种方法是通过消除现有的TAM或通过抑制进一步的TAM募集来减少或消耗TAM,这种策略主要通过定位(i)集落刺激因子1 (CSF1)/CSF1 受体 (CSF1R) 信号通路,(ii)趋化因子/趋化因子受体轴,例如CCL2/CCR2、CCL5/CCR5,(iii)IL-8/CXCR2或(iv)CXCL12 /CXCR4轴实现。
第二种方法是通过抑制PI3Kγ信号通路,通过TLR激动剂触发炎症激活Toll 样受体(TLR:TLR3、TLR4、TLR7/8和TLR9)信号通路,将TAM重新极化为M1样表型,或者通过使用激动性抗体CD40抗体实现。也可以通过阻断被称为“don’t eat me”信号的抗吞噬表面蛋白(如SIRPα 或 Siglec-10)来促进TAM的抗原呈递和吞噬作用,同时抗体阻断癌细胞上表达的CD47或CD24。
第三种靶向TAM的分子是TGF-β,它是一种抗炎细胞因子,通常在损伤消退期间由巨噬细胞表达。巨噬细胞既是TGF的来源也是靶点,通过促进额外TGF-β的分泌,引起TAM的正反馈回路并维持免疫抑制性TME。通过TGF-β阻断靶向TAM已经被使用,也可与STING 激动剂或抗PDL1阻断联合使用,并在临床前模型中显示肿瘤消退。
第四种方法就是增强肿瘤细胞吞噬作用,最新的研究强调了多种抗原靶向的重要性,它既可以提高CAR-T细胞治疗的有效性,又可以减少脱靶反应,例如生成具有靶向CD47和TAG-72的两个串联CAR的CAR-T细胞。双重靶向策略使CAR-T细胞破坏表达低抗原水平的肿瘤细胞的能力增强,有利于增加串联CAR与肿瘤细胞的亲和力。有的研究者研究发现经工程改造的NanoCAR-T 细胞可分泌抗CD47纳米抗体能够抑制肿瘤生长,同时避免全身抗CD47治疗遇到的**。与标准CAR-T细胞相比,这种TAM重编程策略显示出优异的抗肿瘤活性。
➣髓源性抑制细胞
MDSC是具有免疫抑制性未成熟的骨髓细胞。根据形态和细胞表面抑制物分为两种亚型:单核细胞MDSC (M-MDSC) 和多形核/粒细胞MDSC (PMN-MDSC)。它们通过诱导Tregs和产生精氨酸酶(ARG1)、诱导型NOS(iNOS)和抑制性细胞因子如 IL-10 和 TGF-β,有助于产生肿瘤免疫抑制环境。目前增加CAR-T细胞对MDSC免疫抑制作用抵抗的策略:(i)防止 MDSC 分化和募集到肿瘤床;(2)消耗肿瘤浸润性MDSC或(3)减轻MDSC的免疫抑制作用。
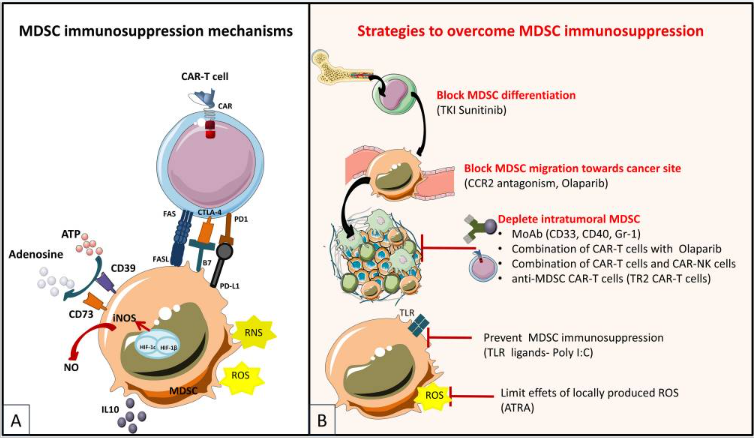
图6
MDSC在肿瘤微环境中发挥免疫抑制机制(A),
克服 MDSC 诱导的免疫抑制的策略(B)
D 免疫检查点分子和免疫抑制介质的表达
免疫检查点(IC)通过调节免疫反应的时间进程和强度,确保维持免疫稳态。然而,来自IC的基于受体的信号级联在T细胞中发挥负调节作用,通过诱导免疫耐受,从而使肿瘤逃避免疫监视。第一个被确定为T细胞和CAR-T细胞抑制和凋亡的重要受体的主要IC是CTLA-4和PD-1。在癌症中广泛研究的其他免疫受体还有 LAG-3、TIGIT、T细胞免疫球蛋白和含有蛋白3 (TIM3) 的粘蛋白以及B和T淋巴细胞衰减剂 (BTLA)。由于PD1/PD-L1抑制是研究最多的轴,我们来主要关注一下CAR-T细胞治疗中的PD-1/PD-L1抑制。
PD-1是B7/CD28家族的成员,它通过与两种配体PD-L1和PD-L2相互作用发挥调节T细胞活性的作用。PD-1/PD-L1结合阻碍IFN-γ和IL-2的合成,从而降低T细胞增殖。为了防止TME中的CAR-T细胞衰竭和免疫抑制可以使用不同的策略,比如:(i)CAR-T细胞与免疫检查点抑制剂(ICI,如抗 PD1 或 PD-L1抗体)的组合;(ii)PD1 介导的抑制也可以通过设计分泌 PD-1 阻断或 PD-L1阻断 单链可变片段(scFv)的 CAR-T 细胞来克服;(iii)设计基因修饰的CAR-T细胞,该细胞表达显性负PD-1受体 (PD-1 DNR),干扰PD1下游信号传导或PD-1嵌合开关受体(CSR),将抑制信号转变成激活信号。(iv)最后一种策略通过基因敲除或通过shRNA(短发夹 RNA)抑制使PD1表达缺失。
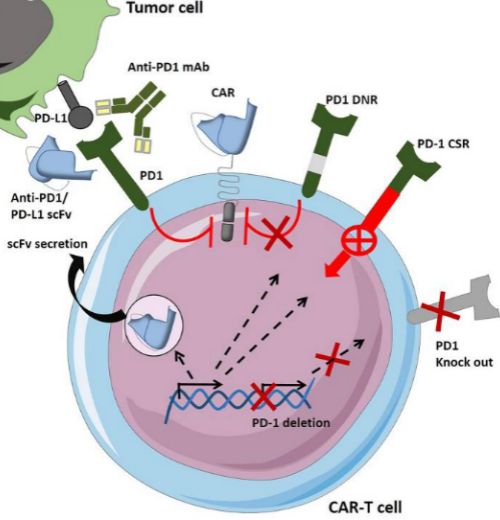
图7 CAR-T 细胞克服负性免疫检查点调节抑制作用的策略(PD1/PD-L1为例)
总之:肿瘤血管系统和细胞外基质等恶性组织的特定性阻碍了CAR-T细胞的渗透,另一方面,调节细胞的存在、免疫检查点的过分表达和TME的缺氧条件会耗尽过继转移的T细胞削弱其抗肿瘤活性。目前所有针对实体瘤的CAR-T还处于探索阶段,并面临着很多的挑战。期待着早日攻克这些难题。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























