
肿瘤的发生、发展与遗传物质密切相关。研究表明,即使核苷酸序列没有变化,仍然有可能发生基因表达差异,进而导致疾病的发生。这种调节机制属于表观遗传学范畴,表观遗传学调控主要有DNA甲基化、组蛋白修饰以及微小RNA转录后调控。DNA附着于组蛋白核小体,其转录的开启和关闭受组蛋白修饰状态调节。组蛋白去甲基化酶1(lysine specific demethylase1,LSD1)可以调控组蛋白修饰状态,其表达异常与众多肿瘤疾病的发生和预后有密切关系。
一.组蛋白去甲基化酶1(LSD1)与肿瘤
组蛋白去甲基化酶1(LSD1),又被称作KDM1A/AOF2,是首 个被发现得组蛋白特异性去甲基化酶。LSD1催化黄素腺嘌呤二核苷酸(FAD),属于单胺氧化酶家族。研究显示,LSD1在人、鼠、昆虫和线虫等种属中均有表达,但结构均十分保守。在LSD1被发现之前,组蛋白甲基化修饰长期被认为是不可逆的,直到LSD1被发现,才了解组蛋白甲基化修饰受到组蛋白甲基化酶和去甲基化酶共同调节,保持动态平衡。
图一 组蛋白去甲基化酶1(LSD1)
从结构来看,LSD1包括N端的SWIRM结构域,C端的AOL(胺基氧化酶)结构域和中心定位的Tower结构域。LSD1可以特异性地去除组蛋白H3第4位赖氨酸(H3K4)和第9位赖氨酸(H3K9)的单、二甲基化,从这调节基因转录(包括激活或抑制),相关过程与肿瘤的发生和发展密切相关。
现有的研究证实,LSD1可以调控肿瘤基因表达,促进肿瘤细胞的增殖、侵袭和转移。LSD1在肺癌、胃癌、结直肠癌、膀胱癌、食管癌、前列腺癌、肝癌和神经胶质瘤、急性粒细胞白血病等多种癌症疾病中均被发现高表达,且表达程度与癌症分级、恶性程度和预后均有关。
二.靶向LSD1的肿瘤治疗策略
自LSD1在2004年被发现以来,研究人员就开始了LSD1靶向治疗策略的研究,目前已在LSD1生物学功能研究、抑制剂开发等方面取得了较大进展(图二)。
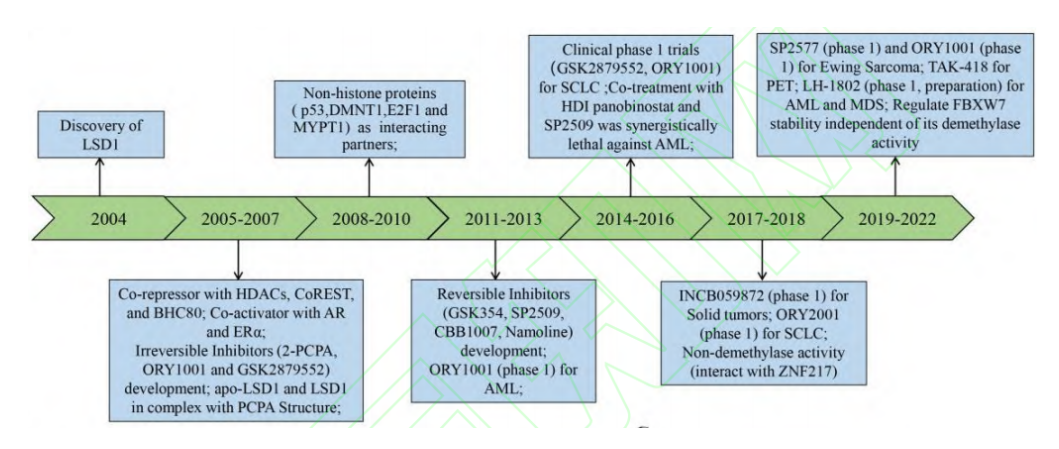
图二 LSD1靶向治疗发展进程
当下,开发高效、低毒的高选择性LSD1抑制剂已经成为药物开发领域的热点。LSD1抑制剂按照作用机制可分作两类:不可逆抑制剂和可逆抑制剂。不可逆抑制剂按照结构主要有苯环丙胺类、炔丙基胺类以及苯乙肼类3类,其都是通过与LSD1的辅因子FAD形成加合物从而抑制LSD1的去甲基化作用。目前已有7种苯环丙胺类LSD1不可逆抑制剂获得美国食品药品监督管理局(Food and Drug Administration,FDA)批准进入临床,包括Tranylcypromine、ORY-1001、ORY-2001、GSK-2879552、IMG-7289、INCB059872 和TAK-418(其中,GSK-2879552、INCB059872以及TAK-418的多项临床试验因为安全性、脱靶效应等问题而终止),涉及的癌种包括:急性髓系白血病、小细胞肺癌、骨髓增生异常综合征等。可逆抑制剂方面,LH-1802进入了临床研究准备阶段,CC-90011和SP-2577进入了临床评估阶段。
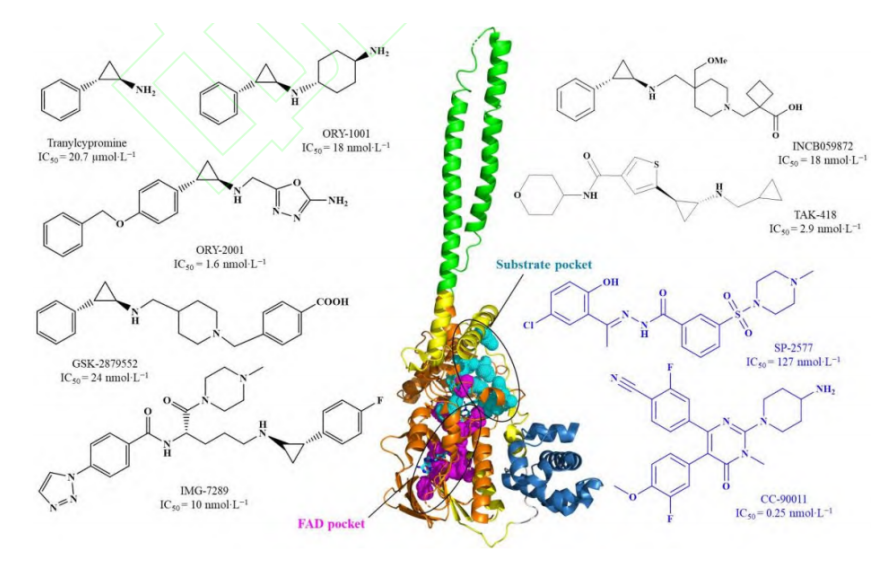
图三 LSD1靶向分子
三.代表LSD1抑制剂
1.MAOs相关抑制剂——Tranylcypromine(TCP)
LSD1属于单胺氧化酶,其催化区域与MAO-A、MAO-B具有同源性。正因如此,早期开发的LSD1抑制剂多与MAO-A和MAO-B有关。Tranylcypromine(trans-2-phenylcyclopropylamine,TCP),对FAO超家族的单胺氧化酶A(MAO-A)和 B(MAO-B)的抑制活性分别为Ki = 102 μmol·L−1和Ki = 16 μmol·L−1,可抑制5-羟色胺和去甲肾上腺素的分解,从而提高大脑中神经递质水平。该药物在1961年被FDA批准用于治疗深度抑郁症。做为MAOs抑制剂,TCP最早证实可抑制LSD1的催化活性。
TCP对FAO超家族的选择性较差,是一种非特异性的LSD1抑制剂。研究显示,抑制LSD1可以增强视黄酸受体抑制剂全反式维甲酸(all-trans-retinoic acid,ATRA)急性髓系白血病(AML)治疗的敏感性。2014年,马丁路德·哈勒维腾贝格大学发起了一项ATRA与TCP联用的I/II期临床研究(NCT02261779),旨在探讨分析TCP与ATR联用对复发或难治性AML患者的可行性、安全性、有效性。在该研究中,TCP(片剂)10 mg口服/d,ATRA(软胶囊剂)按45 mg·m-2的剂量从第7天开始口服,治疗周期1年。2015年,研发人员一项针对TCP的临床研究(NCT02717884),主要研究TCP对非M3型AML或MDS患者对ATRA致敏性影响的临床I/II期研究。2020年,研究人员又发起了一项评估TCP和ATRA联合治疗复发或难治性AML和MDS的安全性以及耐受性的临床I期研究。临床研究结果显示,LSD1抑制剂TCP可以增强AML细胞对ATRA敏感性,并可能恢复MDS和AML患者对ATRA的敏感性。并且,TCP和ATRA联用显示出了较高的耐受性和安全性。
2.首 个进入临床研究的可逆LSD1抑制剂——CC-90011
CC-90011是首 个进入临床研究的可逆LSD1抑制剂,其对LSD1的半数抑制浓度IC50 = 0.25 nmol·L−1。2021年,生物制药龙头——新基医药/赛尔基因(Celgene Corporation)针对CC-90011开展了一项Ⅰ/Ⅱ期临床研究(NCT04748848),用于评估CC-90011与AML传统疗法(BCL-2抑制剂维奈托克(venetoclax)和胞嘧啶核苷类似物DNMT抑制剂阿扎胞苷(azacitidine))联用治疗AML的安全性、耐受性和疗效。研究显示,CC-90011与AML传统疗法联用可以抑制与AML发病机制相关的异常LSD1活性,增加AML患者对传统疗法的敏感性。
近期,研究人员发起了一项针对CC-90011与广谱抗菌药物利福平(rifampicin)或伊曲康唑(itraconazole)联合用于复发或难治性实体瘤和非霍奇金淋巴瘤的安全性和有效性研究临床实验(NCT02875223)。此外,研究人员针对CC-90011发起了多项目临床研究,包括NCT03850067、NCT04350463、NCT04628988等。已有的临床研究结果显示,CC-90011耐受性好,药代动力学特征优势明显,单用和联用用于肿瘤治疗的前景看好。
小结
LSD1做为一个肿瘤药物的新靶点,自被发现以来,迅速成为了医药研究领域的热点。近年来,LSD1抑制剂研究如火如荼,取得了较大的进展,多款药物成功进入临床研究,并表现出了很大的应用前景。目前,虽然尚未有LSD1抑制剂成功上市,但数据已经证明了LSD1是一个优秀的肿瘤治疗靶点,随着研究的不断深入,LSD1必定在肿瘤治疗上大有作为。
参考文献:
1.Annual review of lysine-specific demethylase 1 (LSD1/KDM1A) inhibitors in 2021,2022;
2.LSD1 在肿瘤中的研究进展及其抑制剂的开发现状,2020;
3.组蛋白去甲基化酶 LSD1 抑制剂临床研究进展,2022.
作者简介:云天,药物化学博士,主要从事小分子药物研究,尤其擅长小分子药物的合成工艺及后期药物开发研究,已完成多个抗癌药物分子的合成和活性评估。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57






















