转录因子介绍
转录因子通过与特定DNA 结合序列结合来帮助打开或关闭基因,调控了几乎整个基因组。所有转录因子 (TF) 都包含两个通用蛋白质结构域:一个与特定 DNA序列结合的 DNA 结合域 (DBD),一个招募共激活因子的效应域。许多转录因子还包含一个或多个调节结构域,这些结构域通常用于调节转录因子定位和功能,例如STAT3,通过其SH2结构域来实现二聚化并调控靶基因的表达。
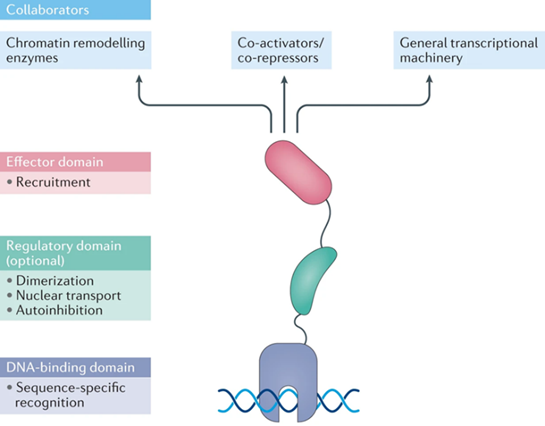
据估计,人类基因组中至少有 1,600 个 TF,其中约 19% 与疾病表型相关。如 p53、Myc、雄激素受体 (AR)、雌激素受体 (ER) 、缺氧诱导因子 (HIF)、NF-κB等转录因子,它们参与各种促癌事件,p53 是研究最多的肿瘤抑制因子,在超过 50% 的人类癌症中发生功能获得性突变失活或变成致癌物。核受体 AR 和 ER 分别与内分泌驱动的前列腺癌和女性癌症(如乳腺癌、卵巢癌和子宫内膜癌)的发病机制密切相关。HIF-1在大多数实体瘤中过度表达。NF-κB是经典激活巨噬细胞的关键转录因子,可介导产生大量炎症因子。
转录因子在选择性基因调控中起基础作用,因此比激酶等上游信号蛋白具有更高特异性的疾病调节能力,并且由于基因靶标受DNA 结合特异性控制,所以单个 TF 通常只调节有限的一组基因靶标,因此这种抑制剂也不容易其他药物常见的补偿性耐药机制。所以靶向异常的转录因子是一种有效治疗疾病的策略。
但由于转录因子的结构异质性,并缺乏活性位点,传统上认为其难以成药。研究表明转录因子存在高度动态的蛋白质-DNA 和蛋白质-蛋白质相互作用,蛋白质-DNA 相互作用界面通常是凸面且带正电荷,而蛋白质-蛋白质相互作用界面通常更平坦且缺乏结合袋,小分子很难直接靶向。
靶向转录因子的小分子抑制剂
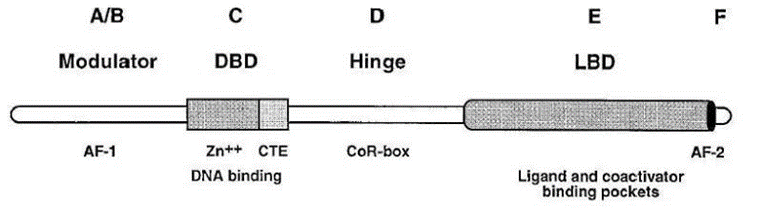
其中,核受体 (NRs)和bHLH-PAS家族是类特殊的 TF 家族,其具有一个 DNA 结合域 (DBD) 和一个配体结合域 (LBD),bHLH-PAS家族是通过与其家族成员二聚化形成LBD。当与小分子配体结合时,会诱导 NRs 构象变化,调节它们与各种转录辅助调节因子(如共激活因子或辅助抑制因子)的相互作用,从而控制下游靶基因的表达,目前开发的小分子抑制剂主要靶向这两类TFs。
核受体包括雄激素AR、雌激素ER等。AR与 AR LBD 的结合导致细胞核易位并启动靶基因表达,这是前列腺癌的关键驱动因素,因此 AR 是一种重要的治疗靶点。AR拮抗剂与LBD结合会阻断AR信号通路,恩杂鲁胺、阿帕鲁胺和darolutamide是第二代AR拮抗剂,最近已被FDA批准用于治疗人类前列腺癌,与第一代相比疗效更强,副作用减少。此外,一些AR拮抗剂正在进行临床试验,如proxalutamide和EPI-7386。为避免不良副作用(如AR信号通路对肌肉和骨骼的合成发挥代谢作用),在过去的二十年中合成和开发了选择性AR调节剂(SARMs),如GTx-024,AR或 SARM 与 AR LBD 结合并诱导构象变化,从而促进募集共激活因子,并激活基因表达。
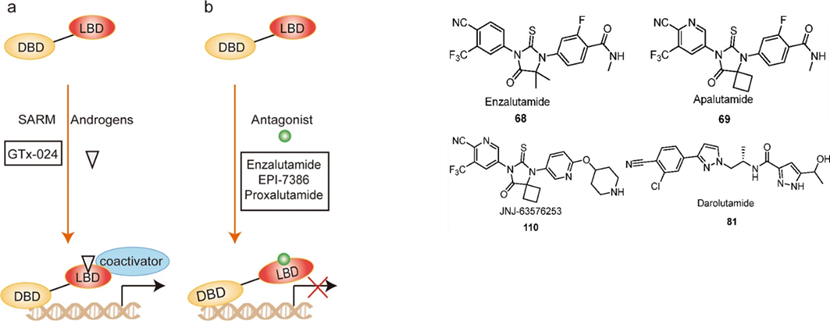
大约 75% 的乳腺癌都是 ER 阳性的, ER 信号传导是乳腺癌的关键驱动因素。选择性ER调节剂SERM(如他莫昔芬)与 LBD 结合,阻断募集共激活因子所需的构象变化,从而降低靶基因表达。

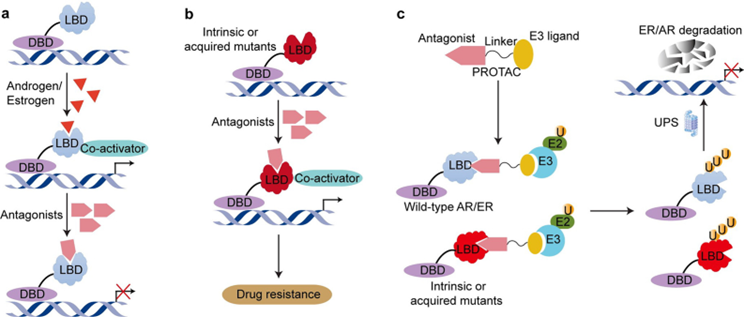
尽管AR或ER的抑制剂或选择性调节剂能降低靶基因表达,但内在和获得性抗性的出现限制其应用。以事件驱动的方式诱导靶蛋白质降解的 PROTAC 技术在克服这个问题方面显示出巨大前景,将在下文介绍。
最后,尽管 NR 由于LBD的存在最易成药,但靶向 NR所有家族成员仍然是一项非常具有挑战性的任务。例如那些被称为孤儿受体的 NR,其没有指定的内源性配体或明显的配体结合口袋。此外在某些疾病中, NR 剪接变体的出现会使传统药物失效,例如 AR-V7,为 AR 的剪接变体。传统的 NR 靶向药物都是与其 LBD 口袋结合。最近,NR 共激活因子结合抑制剂也被开发用于调节 NR 活性。
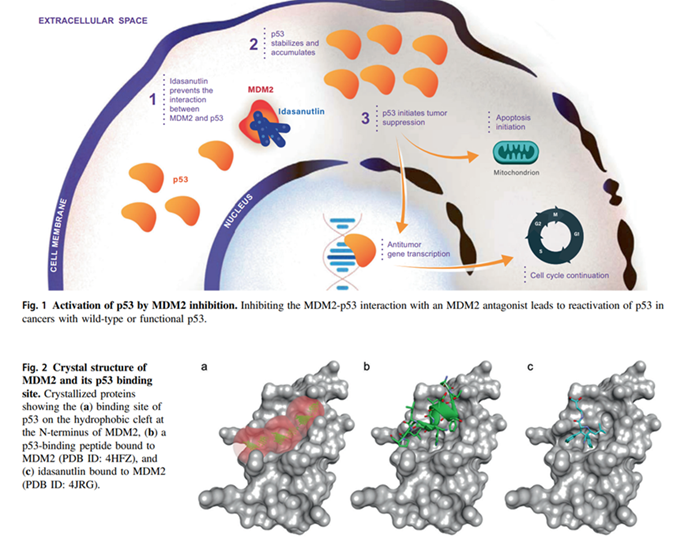
蛋白质-蛋白质相互作用 (PPI) 在许多生理过程中发挥重要作用,并且在疾病中经常失调。因此,PPI 也被认为是潜在的治疗靶点。例如p53-MDM2抑制剂和menin-MLL抑制剂。在癌症中,TF p53(“基因组的守护者”)和泛素 E3 连接酶 MDM2 之间的 PPI 通常作为癌细胞通过泛素-蛋白酶体系统下调 p53 水平来逃避凋亡的机制。然而p53 是一种高度无序的 TF,因此开发一种与 p53 结合并使其免受蛋白酶体降解制的小分子极具挑战性。幸运的是MDM2 的 p53 结合位点是一个相对较小且定义明确的疏水口袋,因此可以设计抑制它们相互作用的小分子药物,如HDM201 和 RG7388来抑制p53的泛素化。
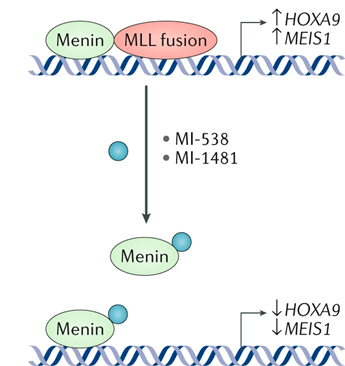
共激活因子menin与MLL融合蛋白的相互作用对于 MLL 融合阳性白血病是必不可少的,因此开发小分子抑制剂对治疗白血病有效,例如已经开发的MI-538 和 MI-1481,通过阻断其相互作用而抑制白血病。
但蛋白质-蛋白质界面通常是疏水性的,难以开发具有理想成药性质的小分子化合物。与酶的活性位点或受体的结合位点相比,蛋白质-蛋白质相互作用界面更加平坦稳定,难以靶向。此外,蛋白质-蛋白质界面相对没有特征,缺乏可以完全容纳小分子配体的疏水腔。因此蛋白质-蛋白质相互作用抑制剂的未来发展方向应该是优先设计新的化学、生物和计算工具,并且扩大化学库以筛选有效小分子。
靶向转录因子的PROTAC降解剂
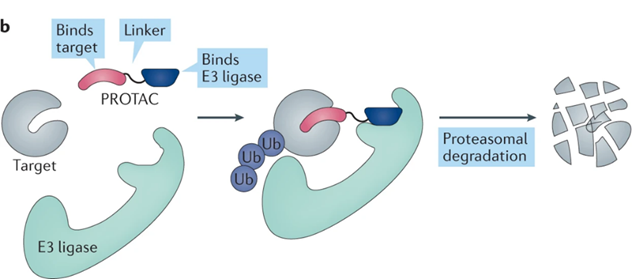
在细胞中,必须破坏不必要或错误折叠的蛋白质以确保细胞蛋白质稳态,这个过程受到细胞内泛素蛋白酶体机制的严格控制。通过泛素-蛋白酶体系统劫持内源性 E3 连接酶降解感兴趣的蛋白质 (POI),开发了靶向蛋白水解的嵌合体 (PROTAC)。PROTAC 是一种异双功能分子,含有一个与 POI 结合的配体和一个 E3 泛素连接酶的共价连接配体,通过泛素-蛋白酶体系统导致蛋白质靶标的降解。
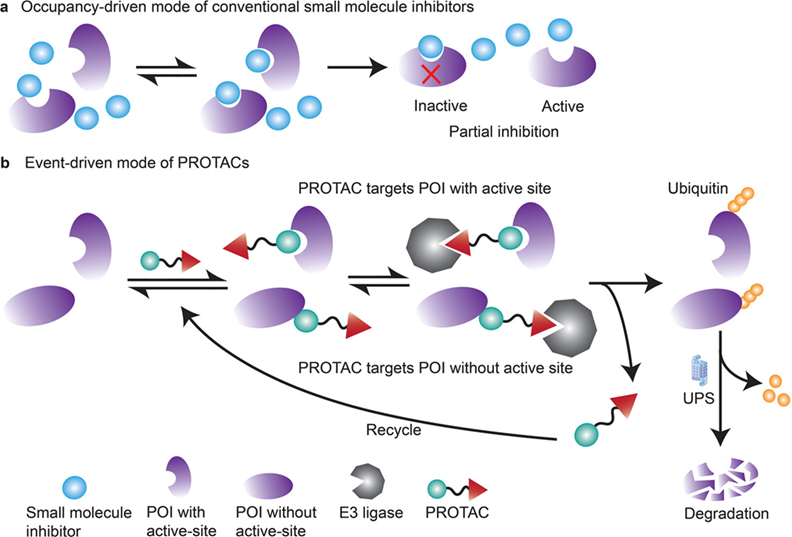
与传统 SMI 的占用驱动操作模式不同,PROTAC 以事件驱动的方式降解POI,无需分离配体来识别靶向 POI ,提供了一种新的策略来抑制以前无法成药的靶标,且具有较强的靶标选择性,并有可能克服传统 SMI 常见的耐药性。ROTACs 能消除靶蛋白的所有功能,而不仅仅是特定小分子抑制剂所靶向的活性。并且这种方法也是催化的,允许单个分子通过重复的结合和降解循环介导多个靶蛋白的破坏,可以减少给药频率。但也有一些局限性,如不良的药代动力学特征、严重依赖E3泛素连接酶和存在潜在脱靶毒 性。
目前靶向AR的PROTAC研究很多。2008 年,Crews 实验室报告了第一个 PROTAC AR 降解剂,该降解剂使用 MDM2 抑制剂作为 E3 连接酶配体,比卡鲁胺作为 AR 拮抗剂。虽然化合物131仅在微摩尔浓度下降解细胞中的 AR 蛋白,但它提供了重要的概念验证。
化合物135-137用高效的AR拮抗剂和VHL配体设计PROTAC,尽管都具有出色的降解能力,但在动物体内的口服生物利用度较低,这限制了其进一步发展。ARD69 和 ARD266 表明,具有高刚性接头的 PROTAC 能提高活性。
由高效的 AR 配体和带有刚性接头的 CRBN 配体设计的 AR PROTAC 已显示出优异的 AR 降解效力、有效的细胞生长抑制活性、良好的 PK 曲线以及有效的抗肿瘤功效。目前ARV-110已进入临床二期,它由强效抑制剂,CRBN配体通过刚性连接子相连,具有非常出色的AR降解效力、在野生型和具有 AR T878A 和 H875Y 突变体的前列腺癌患者中效果明显。Avinas 还有一款口服PROTAC 药物ARV-766正在进行临床一期试验。
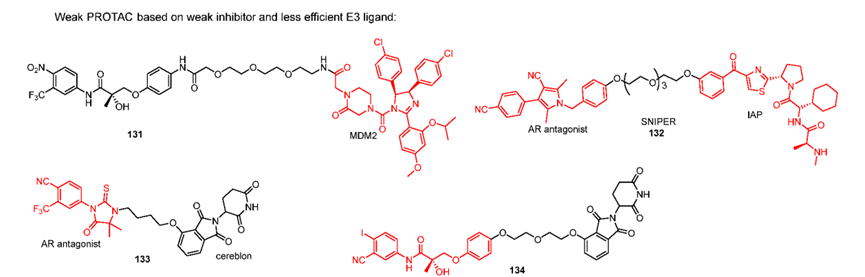
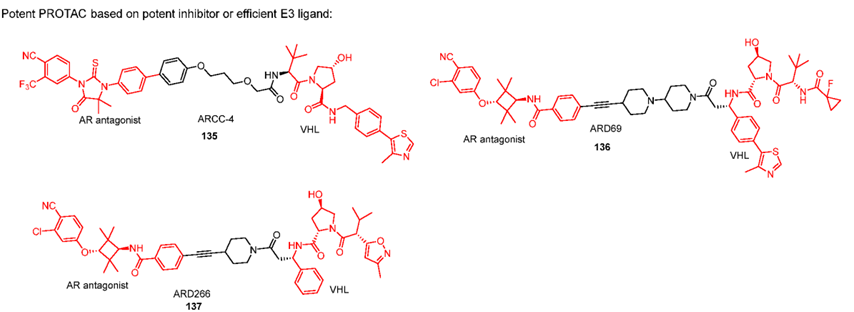
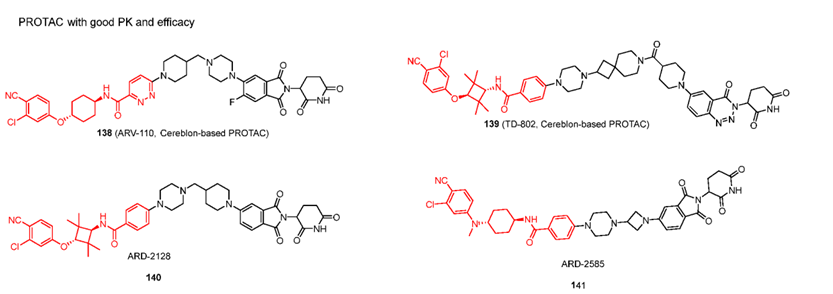
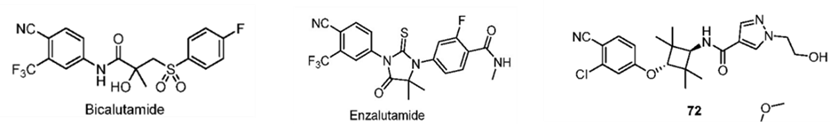
尽管目前已研发出大量的激动剂和拮抗剂,但报道的 AR PROTAC 仅使用少数 AR 抑制剂及其衍生物,包括比卡鲁胺、恩杂鲁胺和化合物72来自辉瑞,且只有 CRBN 配体才能提供良好的口服生物利用度。
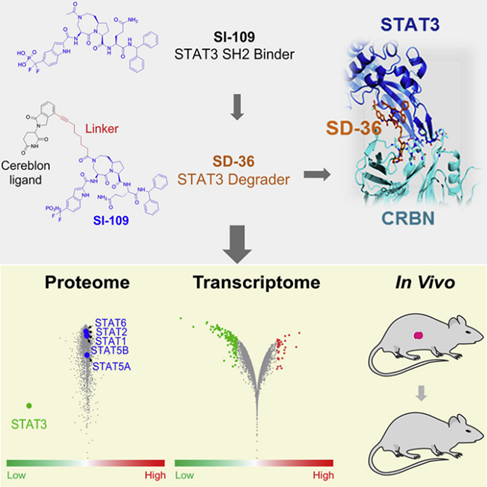
信号转导和转录激活因子 3 (STAT3) 是 STAT 家族的成员,可响应各种细胞因子、生长因子和癌基因信号而被激活,其激活通常与预后不良有关。STAT3通过一个Tyr705位点磷酸化的单体与另一个单体的SH2结合域的结合口袋相互作用进行二聚化,随后激活靶基因。因此STAT3 SH2 结构域的抑制剂能抑制STAT3活性。但 STAT3 和其他 STAT 家族成员共享一个高度结构同源的 SH2 结构域,难以获得高度选择性的 STAT3 抑制剂。并且单体 STAT3 蛋白也具有转录活性,因此阻断 STAT3 二聚化的 STAT3 SH2 结构域抑制剂仅部分抑制 STAT3 的基因转录活性。研究人员通过优化以前的 STAT3 SH2 结构域抑制剂 CJ-887,开发了一种可渗透细胞的 高效STAT3 降解剂SD-36。
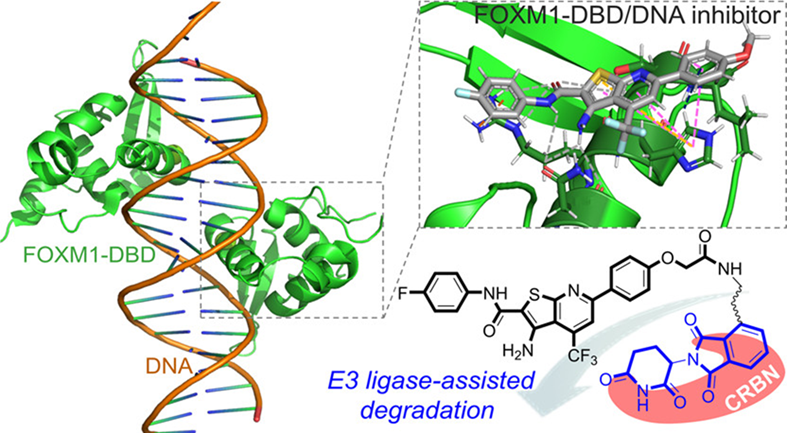
Forkhead box M1 (FOXM1),是一种对细胞分裂至关重要的转录因子,FOXM1 转录活性增加与癌症患者的不良预后有关。对FOXM1 的 DNA 结合域进行计算机建模,确定了一种合适的抑制 FOXM1-DNA 相互作用的小分子,并设计了一种基于 CRBN 的 PROTAC,可显着降解 FOXM1 蛋白并具有抗肿瘤活性。
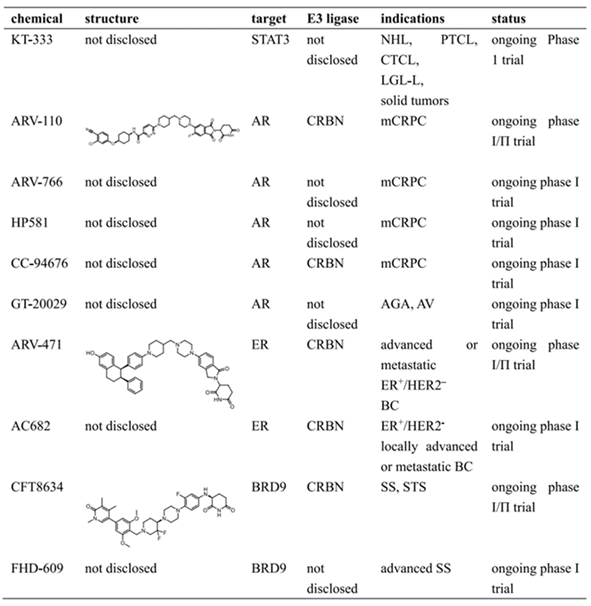
与经典的转录活性抑制剂相比,这些基于小分子的 PROTAC 具有改善的口服生物利用度、低毒 性和卓越疗效等优点,目前这些针对转录因子的PROTACs正处于I期和II期临床试验中。
如上所述,SMI 可以靶向 AR、ER 和 STAT3 等一些转录因子,从而可以基于这些 SMI 设计开发 PROTAC。然而,大多数转录因子仍然无法成药或难以成药。转录因子以序列特异性方式识别启动子和增强子 DNA 中的顺式调节元件,但这些关键功能位点也难以设计SMI。利用转录因子具有特定 DNA 结合序列这一特点,目前几项研究已使用这些寡核苷酸序列作为配体,开发 PROTACs 用于靶向降解转录因子。
Crews开发了一种 TRAFTAC(靶向转录因子的嵌合体),它使用异双功能双链 DNA/CRISPR-RNA 嵌合体同时结合感兴趣的转录因子 (TOI) 和 dCas9-HaloTag7 融合蛋白 (dCas9-HT7,用作中间蛋白) 。CRISPR-RNA 与异位表达的 dCas9-HT7 结合,后者又在 haloPROTAC 存在下招募 VHL E3 连接酶复合物。然后嵌合体的双链 DNA 部分识别 TOI 并使 TOI 和 E3 复合物接近诱导TOI 降解。作为概念验证,他们首先选择了典型的癌基因 NF-κB 作为靶点。NF-κB-TRAFTAC 与 dCas9-HT7 融合蛋白和 NF-κB 亚基 p50 结合,在 haloPROTAC 处理后诱导 p50 显著降解。
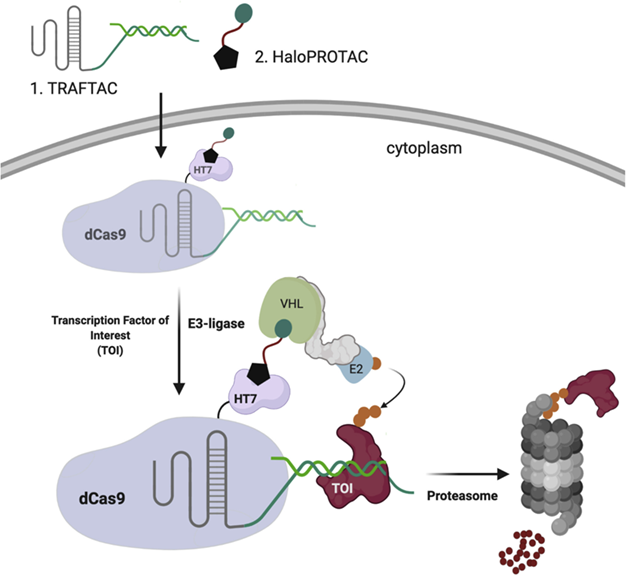
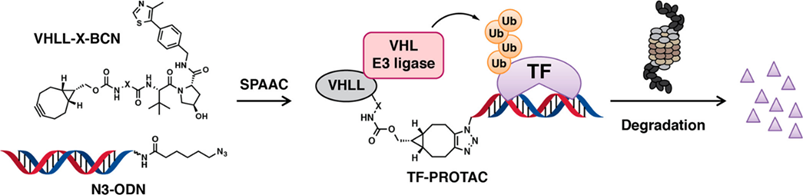
为了开发一种更直接的方法来降解转录因子,研究人员通过点击反应将 DNA 寡核苷酸链与 E3 连接酶配体连接,以选择性降解TOI,称为 TF-PROTAC。
通过无金属铜响应的叠氮化物-炔烃环加成 (SPAAC) 反应将市售的叠氮化物修饰的 DNA 寡聚物 (N3-ODN) 与具有各种接头 (VHLL-X-BCN) 的双环辛炔 (BCN) 修饰的 VHL 配体结合。在去除多余配体 VHLL-X-BCN 的简单纯化过程后,TF-PROTAC 已准备好转染到细胞中,通过 DNA 寡核苷酸链的选择性识别并结合特定TF,使其降解。
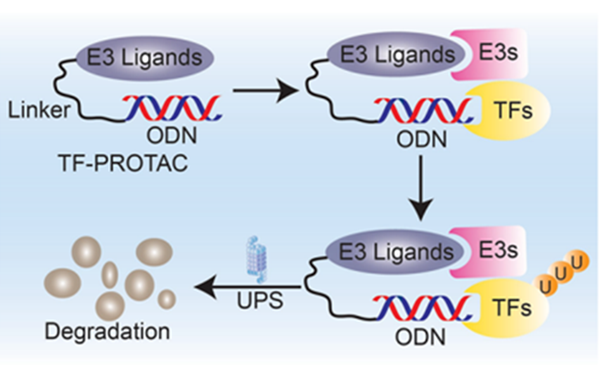
目前开发了两个基于 VHL 的 TF-PROTAC,NF-κB-PROTAC (dNF-κB) 和 E2F-PROTAC (dE2F),分别有效降解细胞中的内源性 p65 和 E2F1 蛋白,并在细胞中显示出优异的抗增殖活性。总的来说, TF-PROTACs 提供了一个可通用的平台来实现 TFs 的选择性降解,但因为缺少体内数据,其临床应用仍有待确定。
总结
尽管基于小分子的 PROTAC 在临床上已显示巨大的前景,但寻找合适的配体来选择性地靶向转录因子依然挑战重重。由于寡核苷酸 DNA 是转录因子的天然配体,基于寡核苷酸的 PROTAC 具有普遍性和灵活性,并且可以通过替换嵌合寡核苷酸的 DNA 序列来靶向其他转录因子,从而为靶向传统上不可成药的转录因子铺平道路。此外,鉴于寡核苷酸具有稳定性、组织渗透性,免疫原性和低毒 性等特点,基于寡核苷酸的 PROTAC 平台为临床开发治疗性PROTAC提供广泛前景。
尽管如此,它们的临床前和临床开发也存在众多挑战,如寡核苷酸的 PROTAC 总是带有大量负电荷,并且其次优的药代动力学和相对较低的药效可能会限制其未来发展。
作为该技术的未来拓展,RNA结合蛋白可以通过RNA识别序列形成RNA-蛋白相互作用参与多种生物学过程,包括RNA转录和mRNA翻译,鉴于转录因子和 RNA 结合蛋白之间的相似性,基于寡核苷酸的 PROTAC 为此类蛋白质的靶向降解提供思路。随着技术的成熟,确定基于 DNA 和 RNA 的 PROTAC 的临床应用前景值得期待。
参考文献
1.Nat. Rev. Drug Discovery 2021, 20, 669?688.
2.Microbiol. Immunol. 2010, 349, 25?60.
3.Molecules. 2020; 25: 2448.
4.Cell 95, 927–937 (1998).
5.Nat Rev Cancer. 2015; 15: 701–11.
6.Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 8554?8559.
7.J. Med. Chem. 2022, 65, 8772?8797
8.Cancer Cell 36, 2019 498–511.
9.J. Med. Chem. 2021, 64, 17098?17114
10.Cell Chemical Biology 2021. 28, 648–661
11.J. Am. Chem. Soc. 2021, 143, 8902?8910
12.J. Med. Chem. 2022, 65, 10183-10194



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























