对于遏制在疾病形成中起关键介导作用的蛋白,药学家通常采用的手段是,合成一个药物分子,通过非共价结合的方式“绑定“它,使其发生空间结构或者其它方面的变化而丧失病理学上的功能,从而实现治愈疾病的最终目的。
然而,设计合适的药物分子抑制剂并不是一件一蹴而就的事情,通常的非共价结合,包括氢键、离子键、偶极-偶极作用(范德华力),疏水作用,芳香环作用等(图1),并不能一劳永逸地锁住蛋白。原因是:吸附-解吸附是一个动态平衡的过程,结合在蛋白上的药物分子会被其它分子取代而被释放,从而失去对目标蛋白致病机制的限制。
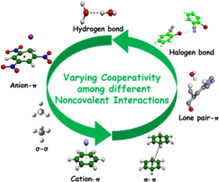
图1. 常见的分子间非共价作用
(图片来源:Chemical Reviews)
基于以上考虑,药学家们采用了更强效的药物分子设计理念——共价键药物。顾名思义,共价键药物与常规非共价键药物的区别在于:共价键药物通过形成与特定的蛋白之间更为牢固的共价键而使其失去致病功能。
共价键是两原子之间通过平均共用电子(相对平均,共用电子对可以偏向电负性更强的原子,从而产生键的极性,但共价还是共价)而产生的新化学键。虽然共价键的形成与裂解也是一个平衡过程,但相对于非共价键来说,一旦产生新的共价键,体系中的低能量是不足以将其裂解的,因而可以相对稳定地存在。
共价键药物与蛋白产生的加合物可以在一定程度上被视为一种不可逆的过程。一旦形成,该蛋白介导的致病过程将被切断,致病过程被终结,直到机体再次生成介导疾病的蛋白,但这通常需要几天的过程。
因此,共价键药物相对于非共价键结合的药物而言,可以产生更长的药效时间,而且所需剂量更低。基于这些优势,共价键药物近年来逐渐成为了制药公司的关注对象。这一点,单从近些年该类药物的专利与文献数量增长便可略知一二(图2)。
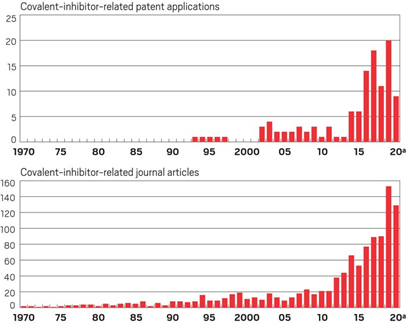
图2. 共价键抑制剂专利与文献数量变化图 (数据来源:CAS)
共价键药物其实并非新兴药物,只是这种理念直到最近20年才被制药行业加以重视并利用。
人们熟知的阿司匹林就属于共价键药物,它的乙酰基在体内与受体酶上的丝氨酸反应,使得丝氨酸被乙酰化,并使该酶失去催化导致炎症和凝血分子形成的功能(图3)。因而阿司匹林在那个时代被视为消炎、解热和抑制血栓形成(从而预防心肌梗死,中风以及心绞痛)的神药。其本质的作用机理都是由于乙酰转移的反应。
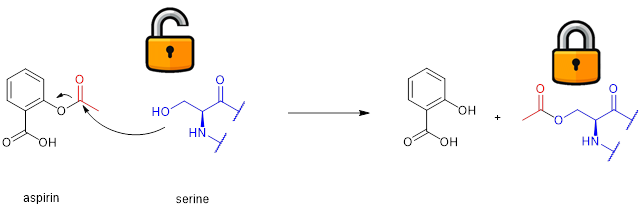
图3. 阿司匹林消炎、抗凝血背后的共价键抑制酶功能的机理
尽管与受体产生共价键的药物,在现代药物产业起源时便已存在,但专属性地研究这类药物却是在一个相对较近的时间范围内。在过去的二十年间,在诸如靶向抗癌药依鲁替尼(共价键结合并抑制BTK布鲁顿酪氨酸激酶)等药物成功案例的基础上,共价键药物得到了快速的发展。
图4展示了药物发展历史长河中著名的共价键药物。其中红色的官能团/化学结构为药物中的活性基团,负责与受体蛋白形成共价键结合而抑制其致病机理。有些人可能会产生这样的疑问:为什么这个药物中会含有高活性的迈克尔加成反应的亲电受体?为什么会有乙氧基这样的高活性结构?为什么会有醛基?它们不会影响药物的稳定性吗?实际上,这些看上去不怎么稳定的药物,可能其治病机理正是通过这些活性官能团与受体蛋白之间形成共价键而实现的。
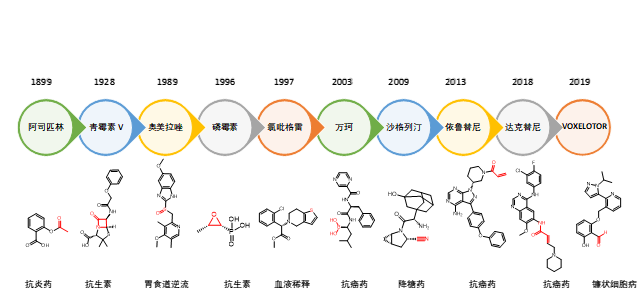
图4. 共价键药物发展历史图
虽然共价键药物有着药效时间长和剂量低的优势,但人们仍然担心这些活性物质会无差别地与体内蛋白结合而引发种种意料不到的副作用,与此同时,这些药物可能引发的免疫响应也是他们关注的内容。因此安全性便成为了共价键药物设计和研发过程的重中之重。
在共价键药物发展的历史上,依鲁替尼被视为具有里程碑意义的上市药物。依鲁替尼以 Imbruvica 的名称销售,它通过共价反应抑制布鲁顿酪氨酸激酶 (BTK),这是一种在某些癌细胞中具有活性的酶 (图5)。依鲁替尼最初是在 2005 年由Celera Genomics 设计的,作为研究 BTK 生物学的工具(可见一开始,依鲁替尼并没有被认真地作为先导药物对待)。Celera 于 2006 年将该化合物作为一揽子交易的一部分出售给 Pharmacyclics,后者才将该分子用于临床开发。
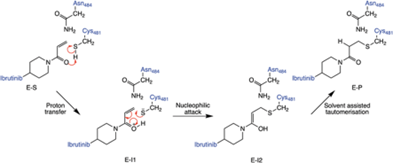
图5. 共价键抑制剂依鲁替尼抑制BTK (布鲁顿酪氨酸激酶) 示意图
由于依鲁替尼一开始被许多制药业从业者忽视,因此它的成功更在行业内引发了思维方式的革命。许多人单从其化学结构便认定依鲁替尼的选择性将会很低,但FDA在 2013 年批准了该化合物用于治疗细胞淋巴瘤,现在它也用于治疗其他五种癌症。这导致人们不得不反省他们当初的判断。
依鲁替尼另一个引人关注的地方在于它创造了极大的利润。它的成功促使艾伯维在 2015 年为 Pharmacyclics 支付了 210 亿美元。依鲁替尼被某些行业内部人士认为是第一个真正验证共价抑制剂价值的神奇分子。
共价键药物分子设计在最近20年也悄然发生变化。在21世纪的最初10年,制药公司在研发共价键药物中采用最多的手段是:在非共价键药物的框架上增添可以与受体蛋白反应的活性官能团,比如丙烯酰胺或者氯乙酰胺。尽管这种手段目前还未完全退出历史舞台,但药物分子设计者已经不必依靠可逆的非共价键药的物改性来发明新的共价键药物。他们可以通过活性片段的数据库来筛选受体结合有效的对象,并以此为基础设计出共价键抑制剂,与那些没有分子结合口袋的受体反应,而这些受体在传统上被视为药物设计的巨大挑战,比如KRAS G12C。
KRAS 是一种参与细胞分裂和增殖的信号传导过程的关键蛋白。但 KRAS 及其家族成员(其突变体存在于 30% 的癌症中)却没有明显的口袋结合部位,药物开发人员花了几十年的时间也没有能够设计出一个满意的非共价结合分子。
2013 年,由加州大学旧金山分校的 Kevan Shokat 领导的一个研究小组发现了KRAS的突变体 KRAS G12C,该蛋白质序列上的第12个氨基酸残基由甘氨酸变为了半胱氨酸。Shokat 发现 KRAS G12C中的半胱氨酸侧链的巯基可以成为药物共价结合的靶点。因为这种甘氨酸突变为半胱氨酸的情况只发生在癌细胞上,因此小分子共价键药物只会特异性地与KRAS G12C作用而不影响常规的KRAS蛋白。
在 KRAS G12C 的情况中,药物分子上的丙烯酰胺亲电基团与半胱氨酸的巯基发生迈克尔加反应。但特殊的是,这个反应似乎利用了蛋白质上半胱氨酸附近的赖氨酸的催化。因为当第13位残基的甘氨酸(第13位置的氨基酸恰好也是甘氨酸)替换为半胱氨酸后,所得到的KRAS G13C变异体对同一丙烯酰胺药物分子没有活性,这或许是Lys的Neigh-bor effect在起作用。在这些数据的鼓舞下,癌细胞中KRAS G12C蛋白上的第12位半胱氨酸成为了药物研发的关注点,引发了共价键药物的研究热潮。
进入临床阶段的KRAS G12C共价抑制剂分子与其受体蛋白之间并没有像传统药物那样的强非共价结合力,但当它们结合在一起时,药物分子上的活性基团迅速与KRAS G12C的Cys12反应。比如Mirati Therapeutics 的临床药物MRTX849 (Adagrasib)(图6)。
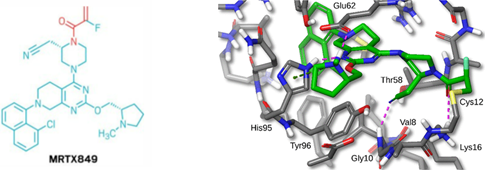
图6. 药物分子MRTX849 (Adagrasib) 上的丙烯酰胺基团(左图红色部分)与KRAS G12C蛋白的Cys12 (右侧蛋白晶体结构上的黄色部分为Cys上的硫原子) 共价结合后的晶体图。(图片来源:Journal of Medicinal Chemistry)
丙烯酰胺基团是共价键药物常利用的亲电结构,从业者对其在药物研发领域内的特性也研究最透彻。依鲁替尼,上面提到的MRTX849 (Adagrasib), 以及去年上市的治疗KRAS G12C突变晚期或转移性非小细胞肺癌的药物Sotorasib (索托拉西布)的分子中都无一例外地包含了丙烯酰胺结构。
也有一些上市的共价键药物没有利用丙烯酰胺结构,例如万珂,它依靠的自己的硼酸基团与受体蛋白上的苏氨酸之间的反应(图4)。还有抗生素药物磷霉素,它介导的是乙氧基与半胱氨酸的反应(图4)。
在蛋白受体方面,研究范围最深, 利用程度最广的氨基酸当属半胱氨酸。很多上市的共价键药物,都是利用药物分子的亲电基团与蛋白受体的半胱氨酸的巯基之间反应。去质子的巯基具有很高的亲核性,可以与药物分子上的亲电基团,诸如丙烯酰胺,发生很快的反应,因此最受药物研发者的青睐。常见的共价键药物的亲电基团,以及它们对应的受体蛋白质上氨基酸,系统地显示在图7中。
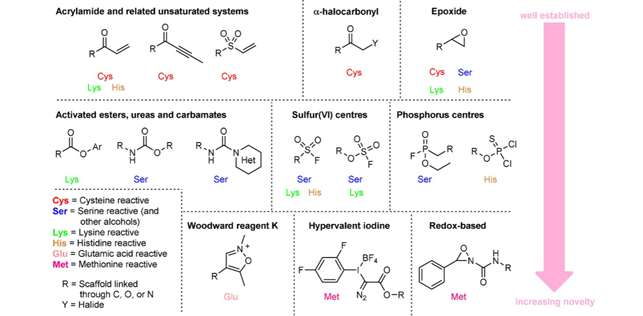
图7. 共价键药物亲电基团与受体蛋白氨基酸对应物(图片来源:ChemBioChem)
方兴未艾的共价键药物虽然在短时间内无法成为药物发展的主流,但它的蓬勃兴起为制药产业提供给了新的强力工具。表1 总结了2010-2019年间FDA批准上市的共价键药物,表2则部分显示了目前处于临床阶段的共价药物。可以想象,随着药物设计的不断发展,共价键类药物如果能在安全性和特异性方面取得突破,必将对制药业产生革命性的影响。
表1. FDA在2011-2019年间批准的共价键药物 (数据来源:RSC Medicinal Chemistry)

参考文献:
[1]Jay B. Fell, et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693.
[2]Samuel E. Dalton, Sebastien Campos. Covalent Small Molecules as Enabling Platforms for Drug Discovery. ChemBioChem, 2020, 21, 1080 –1100.



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























