研究表明,靶蛋白的非酶功能在多种细胞信号通路的调控中起着关键作用,并与许多人类疾病密切相关。然而,传统的小分子抑制剂通常直接靶向催化功能域,通过抑制靶蛋白的酶促功能来发挥作用,而不影响非酶功能。近年来出现的靶向蛋白水解嵌合体PROTAC技术具有同时调控靶蛋白的酶功能和非酶功能的优势,从而为改善小分子抑制剂的不足和探索新的治疗方案提供了潜在的策略。本文总结了针对靶蛋白非酶功能的PROTAC的最新进展,并对未来发展趋势进行了展望。
酶的功能及PROTAC介绍
经典的酶通常由酶结构域和非酶结构域组成。酶的功能域是指酶执行其催化功能的具有特定空间结构的区域。具体来说,酶活性位点包含一个结合区和一个催化区,它们分别参与底物的结合和催化底物发生特定化学反应。而非酶功能区主要通过蛋白−蛋白相互作用(PPI)来调节底物蛋白活性,它独立于催化功能,介导底物与信号通路中不同蛋白成分之间的相互作用,如:变构调控、支架蛋白功能等。
大量研究表明,各种靶蛋白的非酶功能区域参与了细胞分裂、分化、RNA代谢、DNA修复和基因组稳定性的调控。此外,这些非酶的功能与各种人类疾病密切相关,如癌症、心血管病,并在调节细胞信号传导和决定细胞命运中起着关键作用。然而,传统的小分子抑制剂通常直接靶向酶的催化功能域,并通过抑制靶蛋白的酶促功能来发挥治疗效果。然而,这些抑制剂通常不能阻断靶蛋白的非酶功能,并且面临着选择性低、特异性差、临床疗效有限和耐药性等问题。
靶向蛋白降解技术(TPD)被认为是一种富有治疗前景且极具吸引力的治疗策略。PROTAC通常由三部分组成:靶蛋白配体,E3泛素连接酶配体,以及连接链。PROTAC可以招募E3泛素连接酶,通过泛素−蛋白酶体系统(ups)促进靶蛋白的泛素化,进而降解。
与传统的直接抑制靶蛋白的药物开发策略不同,PROTAC通过调节宿主蛋白降解系统来发挥作用。此外,PROTAC可以诱导整个靶蛋白的降解,可以同时阻断靶蛋白的酶功能和非酶功能(图1)。因此,通过PROTAC策略阻断靶蛋白的非酶功能可能是解决传统小分子抑制剂所面临的疾病治疗问题的一种新的和富有治疗前景的策略。
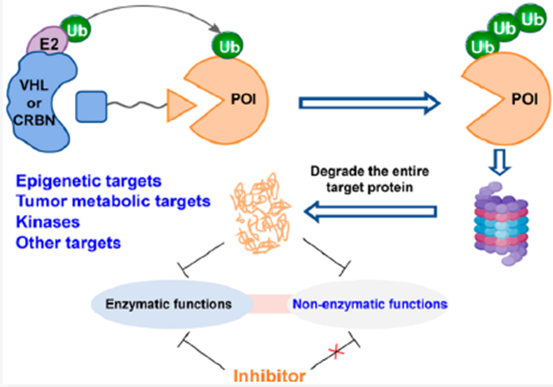
图1.靶向酶结构的PROTAC技术
阻断表观遗传靶标非酶功能的PROTAC
1 靶向EZH2的PROTAC
表观遗传学是研究基因的核苷酸序列在不发生改变的情况下,基因表达的可遗传修饰。表观遗传修饰的异常广泛存在于肿瘤的发生发展过程中,是抗肿瘤药物开发的研究热点。
PRC2是多组合组蛋白中的一员,是表观遗传癌症治疗的重要靶点,具有组蛋白甲基转移酶活性。PRC2复合物由EZH2、EED、SUZ12、RbAp46/48四个主要成员组成。PRC2复合物的过度激活通过沉默肿瘤抑制基因而诱导恶性肿瘤,EZH2是PRC2的一个核心和多功能的催化亚基,在多种癌症类型中过表达,如肺癌、膀胱癌和乳腺癌。EZH2通过甲基化组蛋白H3的27位赖氨酸来抑制肿瘤抑制基因的表达。
近年来,通过直接或间接靶向EZH2的抑制剂的开发取得了重大进展。越来越多的证据表明,EZH2的致癌功能并不完全依赖于其酶活性。除了催化H3K27me和介导与各种细胞过程相关的基因沉默外,EZH2还介导多种癌症中基因的激活,这些基因与EZH2/PRC2的酶促功能无关。
然而,目前所报道的EZH2抑制剂仅通过靶向其组蛋白甲基转移酶活性来下调H3K27me3的水平。由于对EZH2致癌活性的抑制作用不足,EZH2抑制剂的临床疗效有限,且仅对某些癌症有效。因此,迫切需要开发一种新的靶向EZH2的治疗策略。
靶向蛋白降解策略PROTAC为完全阻断EZH2的致癌活性提供了新的机会。
最近,四川大学yu课题组通过将EZH2选择性抑制剂EPZ6438与CRBN配体沙利度胺通过连接链连接,设计合成了EZH2 PROTAC 1(图2)。实验结果表明,化合物1可以完全抑制EZH2的致癌活性。其中,化合物1对PRC2亚基表现出显著的降解效率,72h降解完全。研究发现,化合物1直接与EZH2蛋白结合,而不是SUZ12、EED和RBAP48。因此,化合物1可以将E3泛素连接酶招募到PRC2复合物附近,导致EZH2的泛素化和降解。EZH2介导的间接相互作用诱导了蛋白酶体对其他PRC2亚基的降解,包括EED、SUZ12和RBAP48。然而,由于PRC2亚基的选择性较差,化合物1可能具有潜在的毒 性或副作用。未来对化合物1的研究应集中于如何提高蛋白降解的靶点选择性。
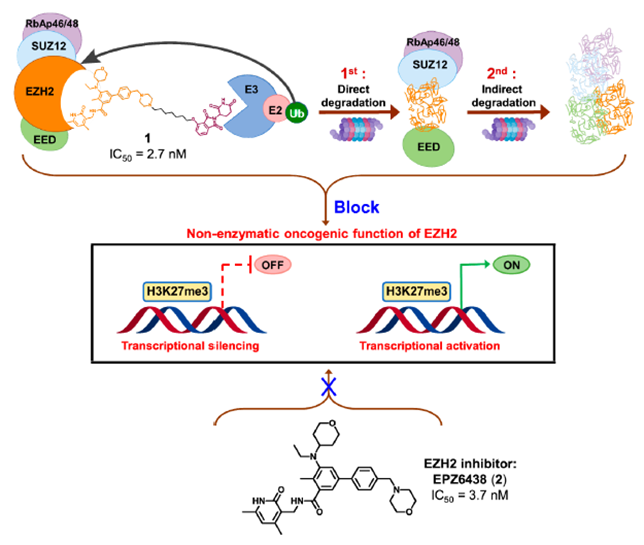
图2.靶向EZH2 PROTAC 1结构
温课题组将选择性EZH2抑制剂EPZ6438和VHL配体通过连接链进行连接,获得了选择性的EZH2降解剂YM281 (3)和YM181 (4)。实验结果表明,这两个EZH2 PROTAC可以靶向淋巴瘤中整个EZH2,通过降解EZH2,进而阻断EZH2的非酶功能(图3)。此外,EZH2 PROTAC显著降低了H3K27me3的水平,并诱导了细胞周期阻滞和凋亡。与此同时,对抑制剂EPZ6438耐药的淋巴瘤细胞系表现出完全的抑制作用,在体内抗肿瘤模型中也表现出较好的抑制效果。
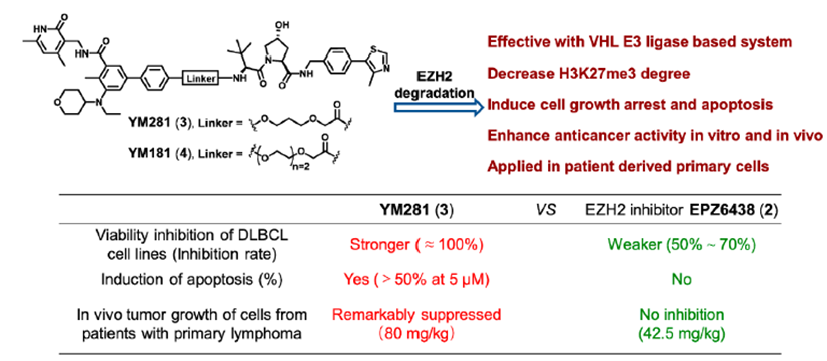
图3. 靶向EZH2 PROTAC YM281 (3)和YM181 (4)的化学结构
由于EZH2-PRC2和EZH2-TAD-cMyc-coactivator这两种EZH2复合物对EZH2介导的促癌功能都非常重要,造成了目前EZH2抑制剂在肿瘤治疗中的局限性。cMyc是一种难以被小分子靶向的致癌因子,它与EZH2有直接的PPI作用,并且独立于PRC2复合物。
因此,研究人员使用PROTAC技术设计了降解剂MS177 (5)(图4),它可以同时针对EZH2的酶功能和非酶功能。实验结果表明,化合物5降解了PRC2复合物成分(包括EZH2、SUZ12、EED),下调了H3K27me3的水平,抑制了依赖于PRC2的传统酶促功能。此外,它还能有效降解cMyc,抑制EZH2的非酶促功能。
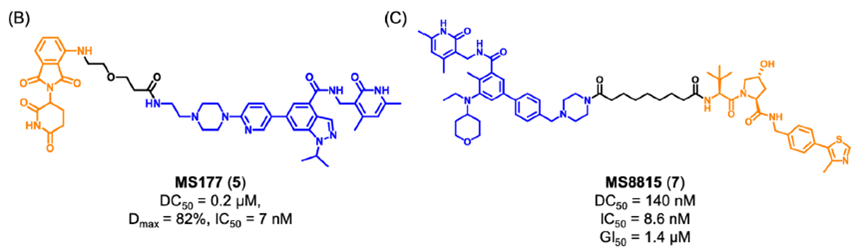
图4. MS177 和MS8815的化学结构
金坚团队开发了一种EZH2选择性降解剂MS8815 (7),以探索其在三阴性乳腺癌(TNBC)细胞中的作用。实验结果表明,化合物7在乳腺癌细胞系MDA-MB-453细胞中实现了EZH2的高效降解(DC50 = 140 nm)。此外,化合物7在多个TNBC细胞系中均具有良好的抗增殖活性。因此,化合物7的开发克服了传统靶向催化位点抑制剂仅针对TNBC细胞中EZH2的催化功能域的局限性。
2 靶向HDAC6的PROTAC
组蛋白去乙酰化酶6(HDAC6)是HDAC家族中的一个微管相关成员,主要位于细胞质中,参与调节错误折叠蛋白的降解、细胞形态和迁移。HDAC6的异常调控与癌症、神经退行性疾病、自身免疫性疾病等多种疾病密切相关。已知的HDAC6选择性抑制剂通过结合HDAC6的C端催化结构域来阻断酶的功能。然而,由于HDAC6中存在多个结构域(图5),如C端泛素结合域(UBD)、N端催化结构域和锌指泛素结合域(ZNF-UBP),目前有多达45个HDAC6抑制剂未能靶向这些功能域。因此,迫切需要新的针对HDAC6的研发策略。
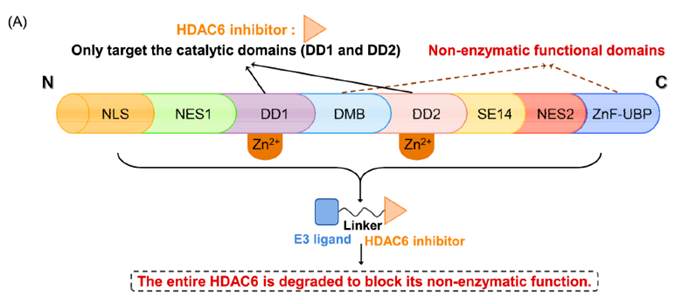
图5. HDAC6 的结构域
目前已经成功报道了多种靶向HDAC6的PROTAC(图6)。其中,化合物9是基于VHL配体的PROTAC,化合物12-14是基于CRBN配体的PROTAC(化合物13和14是阴性对照)。这些PROTAC都表现出显著HDAC6的降解效果和抑制肿瘤细胞生长的效果。这些降解剂通过诱导整个靶蛋白的降解,同时阻断HDAC6的酶促功能和非酶促功能。
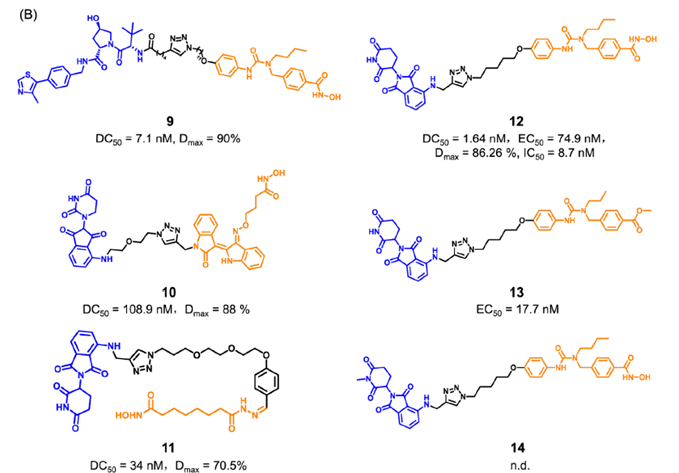
图6.靶向HDAC6 PROTAC化学结构
阻断肿瘤代谢靶点非酶功能的PROTAC
烟酰胺磷酸核糖基转移酶(NAMPT)是烟酰胺腺嘌呤二核苷酸(NAD)生物合成的关键酶,其在肿瘤代谢和炎症中起关键作用。除了在肿瘤细胞的增殖和分化中的功能外,NAMPT还由于其细胞因子样作用而影响免疫微环境。
NAMPT被称为“双侧蛋白”,分为胞内NAMPT(iNAMPT)和胞外NAMPT(eNAMPT)两种类型。研究表明,NAMPT抑制剂仅阻断其酶功能,而不通过eNAMPT调节非酶功能。仅仅抑制NAMPT的酶功能不足以完全抑制NAMPT的致癌功能。由于有限的抗肿瘤功效和剂量依赖性毒 性(例如血小板减少和胃肠道副作用),两种NAMPT抑制剂(FK866和CHS-828)的临床试验停止。因此,迫切需要新的策略来干扰NAMPT的非酶功能。
盛春泉课题组报道了第一个可以降解NAMPT并减少eNAMPT分泌的PROTAC 15(图7), 化合物15通过UPS途径直接降解iNAMPT,从而减少eNAMPT的分泌,促进抗肿瘤免疫,从而阻断NAMPT的酶和非酶功能。在肿瘤小鼠模型中,化合物15也表现出较好的抑制效果。
此外,化合物15能以较低的细胞毒 性和更好的药代动力学特性激活免疫反应。PROTAC 15的开发使我们更好地了解NAMPT的非酶功能在重建免疫抑制肿瘤微环境中的作用,从而促进了NAMPT靶向肿瘤免疫治疗方法的发展。
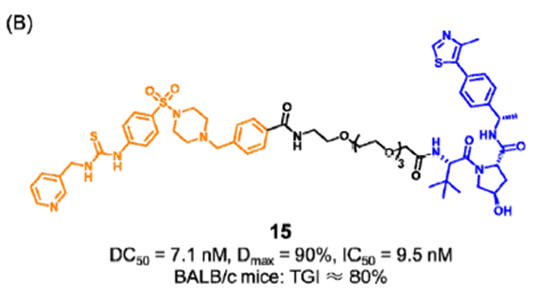
图7. 靶向NAMPT PROTAC 15的化学结构
阻断激酶非酶功能的PROTAC
1 靶向有丝分裂激酶AURORA-A的PROTAC
有丝分裂激酶AURORA-A在有丝分裂过程中对各种蛋白质的磷酸化起着重要作用,其催化活性对整个细胞周期而言至关重要, AURORA-A被认为是抗癌药物发现的重要靶点。然而由于低临床应答率,AURORA-A激酶抑制剂的开发一直停滞不前。研究表明,AURORA-A的非酶功能区可以与MYC家族的原癌基因蛋白结合,使得N-MYC和C-MYC不能被蛋白酶体降解。并且,AURORA-A激酶抑制剂不足以消除AURORA-A的致癌活性。
为了探索AURORA-A激酶的非酶功能,Wolf课题组将AURORA-A临床抑制剂alisertib17与CRBN的E3连接酶配体结合,开发了PROTAC JB170(图8)。研究发现,JB170可诱导AURORA-A的快速、有效和高度特异性降解。AURORA-A的酶活性被认为主要在细胞周期的G2/M期起作用,而其在S期的功能可能与其酶活性无关。作者发现,化合物18可以诱导强烈的S期阻滞,但对G2/M期的细胞聚集没有明显的影响。此外,作者也通过实验证明了化合物18阻滞S期主要由DNA复制过程中AURORA-A的非酶功能引起。
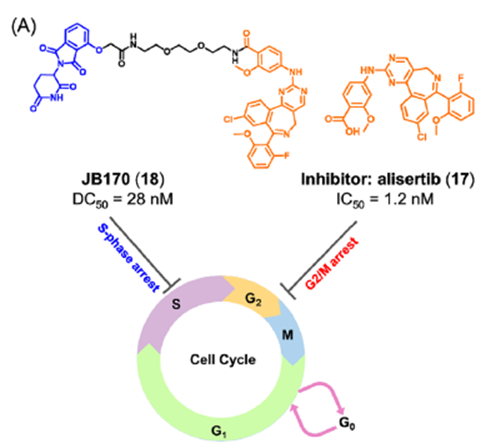
图8. JB170的化学结构
2 靶向FAK的PROTAC
FAK是介导生长因子受体和整合素相关信号转导的细胞质蛋白酪氨酸激酶。FAK主要由三个结构域组成:N-末端FERM结构域、中心激酶结构域和C-末端局部粘附靶向(FAT)结构域(图9)。虽然传统的FAK抑制剂只作用于蛋白激酶结构域以阻断酶功能,但FAK的非酶功能在癌症的发展和进展中也是关键的,并且不能被报道的FAK抑制剂阻断。因此,同时抑制FAK的激酶依赖性酶功能和激酶非依赖性支架功能是抗肿瘤药物开发的新的研究方向。
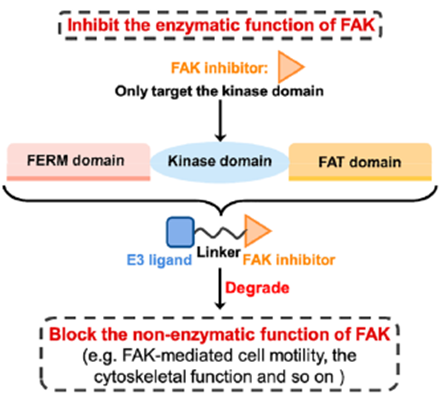
图9.FAK的生物结构特征
耶鲁大学Crews课题组开发了一种靶向FAK PROTAC 19。化合物19通过1,2,3-三唑的聚乙二醇(PEG),将FAK抑制剂defactinib(20)和 VHL配体进行连接而成。PROTAC 19在低纳摩尔浓度下选择性降解(DC50=3.0nM,Dmax=99%)。除影响其激酶依赖性信号活性外,FAK的降解还减弱其激酶依赖性信号。例如,由于FAK介导的细胞运动性主要由激酶非依赖性途径控制,去除FAK显著阻碍了TNBC细胞的迁移和侵袭能力。
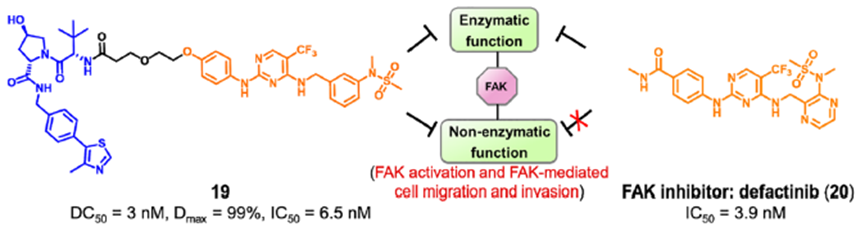
图10. 化合物19的化学结构
清华大学饶燏课题组开发了FAK PROTAC FC-11(图11),并在此基础上进一步研究了FAK的非酶功能。实验结果发现,FC-11可以有效、快速和可逆地降解FAK。同时,FC-11能有效降解小鼠生殖系统中的FAK,并显著下调磷酸化FAK蛋白水平。
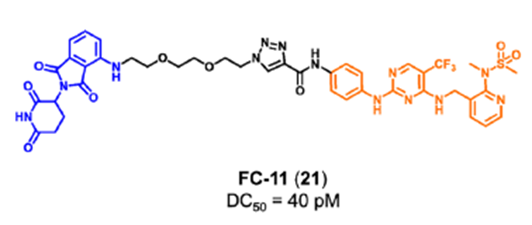
图11.FC-11的化学结构
Law研究团队设计了一种高效、选择性的FAK降解剂GSK215。研究人员将VHL配体和临床FAK抑制剂VS-471通过刚性且短的连接链进行连接而成。化合物GSK215诱导小鼠肝 脏中FAK的持续降解和延长的PK/PD效应(18小时内Dmax为85%,96小时给药后FAK水平降低60%)。值得注意的是,研究人员通过构效关系(SAR)和X射线晶体学分析表明,化合物GSK215具有较高的降解能力源于一种不寻常的短而刚性的连接链,并产生高度协同的三元复合物。
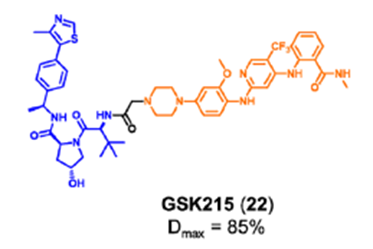
图12. FAK降解剂GSK215的化学结构
针对其他靶点非经典功能的PROTAC
1 靶向FKBP12的PROTAC
肺动脉高压(PAH)是先天性心脏病最常见、最严重的并发症,也是心血管疾病防治的主要问题。近年来的研究发现,铁在多环芳烃的发生和发展中起着至关重要的作用。骨形态发生蛋白(bone morphogenetic protein,BMP)是PAH中的关键蛋白之一,参与调节人体内铁代谢平衡的关键蛋白hepcidin的表达。
FK506结合蛋白12(FKBP12)可结合BMP I型受体并随后抑制hepcidin。FKBP12的这种非经典功能类似于非酶功能,因为它以与催化剂无关的方式调节其与细胞信号途径的不同组分的相互作用。目前报道的免疫抑制剂雷帕霉素和FK506阻断FKBP12与BMP I型受体的结合,从而增加hepcidin。然而,在临床使用中,FKBP12抑制剂表现出免疫抑制副作用。
饶燏课题组报道了PROTAC RC32(图13)。RC32是由雷帕霉素与泊马度胺连接而成。在小鼠模型中,化合物RC32通过激活BMP信号,成功地实现了hepcidin基因表达的上调。与雷帕霉素(24) 或FK506 (25),化合物RC32无免疫抑制活性。这项工作证实了PROTAC介导的FKBP12非经典功能降解治疗低hepcidin相关疾病的可靠性。
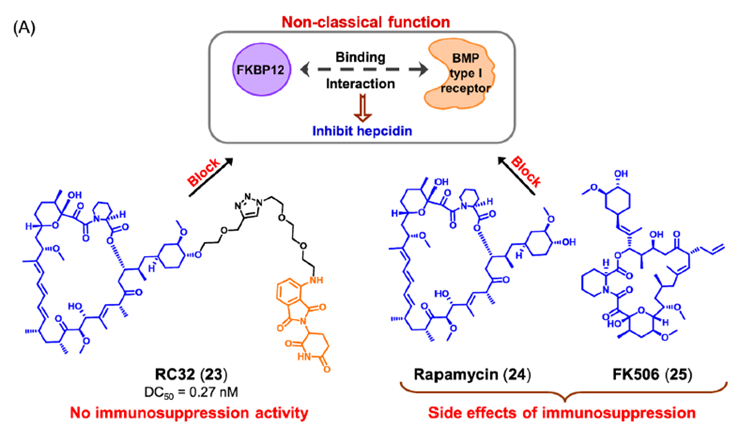
图13. RC32 的化学结构
2 靶向USP7的PROTAC
作为一种重要的抑癌因子,p53是癌症中最频繁突变的基因之一。P53在人类癌症中突变率大于50%。研究发现,突变之后的p53不仅失去对细胞正常生物学功能的调节,而且抑制野生型p53蛋白的功能,从而导致细胞癌变。因此,开发新的药物来治疗p53突变癌症仍然是一个紧迫的问题。然而,p53的突变是相对随机的,这使得开发直接针对p53突变体的靶向药物非常困难。
泛素特异性蛋白酶7 (USP7) 通过去泛素化和稳定MDM2,在调节p53含量中起关键作用。最近,上海药物所周兵课题组设计合成了第一代小分子降解剂U7D-1(图14),U7D-1可有效且选择性地降解USP7(DC50=33 nM)。化合物U7D-1表现出与USP7抑制剂相当或更强的对p53野生型癌细胞生长的抑制活性,尤其是在p53突变癌细胞(Jeko-1细胞,IC)中也显示出显著的抗增殖活性,而USP7抑制剂27显示弱活性。进一步的作用机理研究表明,U7D-1可能通过调节USP7的非酶功能区(凋亡和E2F途径)诱导USP7降解,从而发挥对p53突变癌细胞的抗肿瘤活性。
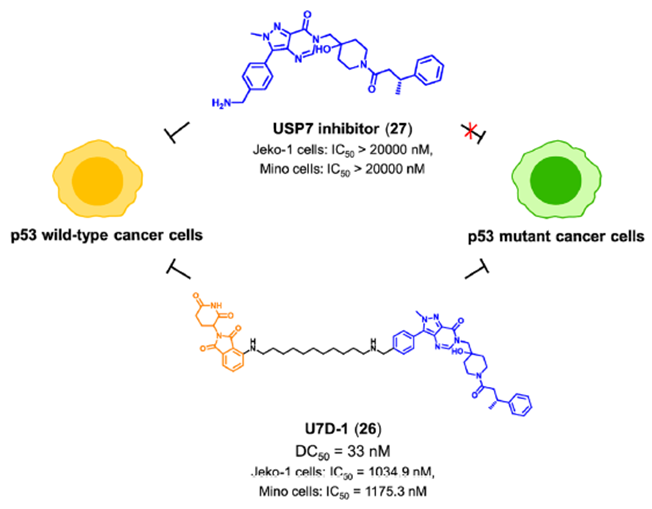
图14. 靶向USP7 PROTAC U7D-1的化学结构
小结
近年来,靶蛋白的非酶功能日益受到关注。由于PROTAC可以同时阻断靶蛋白的酶和非酶功能,且不长时间占据酶结合口袋以诱导整个蛋白的降解的优势而备受瞩目。
目前的研究表明,PROTAC在研究靶蛋白的非酶功能方面显示出良好的优势,并发挥了有效的治疗作用。相比于小分子抑制剂,PROTAC具有更高的靶选择性、更强的疗效、更低的耐药风险和延长的作用。
因此,PROTAC介导的药物靶标的酶和非酶功能的抑制可导致药物活性的显著提高,提供了新的治疗策略,也为研究靶蛋白的非酶功能和相关疾病的机制提供了基础。尽管目前PROTAC对非酶功能的研究还处于起步阶段,还需要更多的研究来发现更多的策略,以调节靶蛋白的非酶功能。但我们依然相信,利用PROTAC阻断蛋白质的非酶功能可能成为药物开发的新方向,前景一片光明。
参考文献
1、Blocking Non-enzymatic Functions by PROTAC-Mediated Targeted Protein Degradation. doi.org/10.1021/acs.jmedchem.2c01159
2、Proteolysis Targeting Chimeras (PROTACs) for Epigenetics Research. Curr. Opin. Chem. Biol. 2020, 57, 8?16.
3、Discovery of a Potent and Selective Degrader for USP7. Angew. Chem., Int. Ed. Engl. 2022, 134 (33), e202204395.
4、Targeting the Non-Catalytic Functions: a New Paradigm for Kinase Drug Discovery? J. Med. Chem. 2022, 65, 1735?1748.
5、Strategies for Designing Proteolysis Targeting Chimaeras (PROTACs). Med. Res. Rev. 2022, 42, 1280?1342.(15) Wang, Y.; Jiang, X.; Feng,



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























