含有SrC同源性2的蛋白酪氨酸磷酸酶2(SHP2)是一种非受体蛋白酪氨酸磷酸酶,主要在多种组织的细胞质中广泛表达,属于蛋白酪氨酸磷酸酶(PTP)家族。
研究表明,SHP2的失调与异常的细胞增殖、分化、粘附迁移和凋亡有关,使得SHP2成为癌症治疗的有潜力的治疗靶点。目前,针对SHP2靶点尚无药物批准上市,多种不同作用模式的SHP2抑制剂已被成功开发,其中一些已经处于临床试验阶段,进展最快的是诺华研发的处于临床二期的TNO-155。
SHP2靶点介绍
1 SHP2结构和功能
SHP2是一种多结构域的酪氨酸磷酸酶,其结构相对保守,由593个氨基酸残基组成。SHP2由两个SH2结构域(N-SH2和C-SH2)、一个保守的PTP结构域和一个灵活的C端尾部组成(图1)。通常,磷酸酪氨酸可以结合SH2结构域以促进PTP结构域与其底物之间的相互作用。
具体来说,N-SH2结构域可以介导SHP2与含磷酸酪氨酸残基的激活剂、去磷酸化底物或适配器蛋白的相互作用。C-SH2结构域与N-SH2和PTP结构域没有直接的表面接触,但可以连接这两个结构域,以提高结合能和底物的特异性。
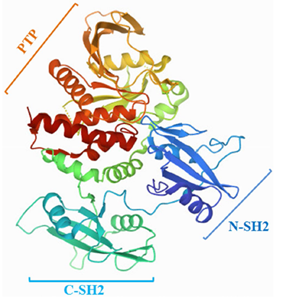
图1.SHP2的蛋白结构
研究发现,SHP2的C端尾部有一个无序的氨基酸残基(残基526-593),其中有两个关键的磷酸化位点:Tyr542,Tyr580和一个富含Pro的基序单元。这些C端酪氨酸激酶(PTKs)磷酸化残基对于SHP2的激活是至关重要的,并为含有磷酸酪氨酸残基的蛋白提供结合位点,如生长因子受体结合蛋白2(GRb2)。SHP2的C端Ser558和Ser562残基对于与含有F-box和WD重复结构域泛素连接酶FBXW7的相互作用也是至关重要的。此外,Lys590是SUMO1的SUMO化位点,介导SHP2与Gab1支架蛋白的相互作用(图2)。
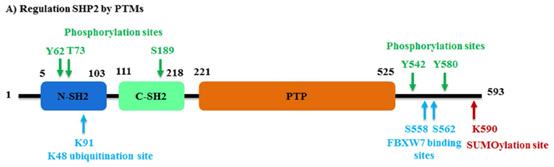
图2. SHP2的翻译后修饰调节功能
在没有N-SH2配体的情况下,由于N-SH2和PTP结构域之间的分子内非共价变构相互作用,抑制SHP2磷酸酶活性。在非活性状态下,Asp61和Tyr62被插入到PTP结构域的催化裂隙中,并封闭其活性位点,这使SHP2保持在一种自抑制状态,并阻断其与含磷酸酪氨酸蛋白结合,抑制磷酸酶活性。因此,N-SH2结构域是SHP2的变构调节因子,并在其激活过程中起着核心作用。
此外,SHP2的激活需要从PTP结构域中去除N-SH2结构域,这需要同时与SH2结构域磷酸化酪氨酸基序结合。研究发现,高亲和力的单磷酸化蛋白也可以通过破坏N-SH2-PTP的相互作用来诱导SHP2的催化活性。据报道,N-SH2的结合裂隙处于柔性状态,N-SH2的中心β-sheet的开放有利于SHP2的激活;而C-SH2结构域主要招募高亲和力的双齿状磷酸肽。与封闭的、自抑制状态的相比,SHP2在开放的、活跃的构象状态下对磷酸化的底物蛋白具有更高的亲和力。此外,SHP2磷酸酶活性也可以通过去除N-SH2或两个SH2结构域来调控。
2 SHP2介导的信号通路
含srC同源-2的蛋白酪氨酸磷酸酶2(SHP2),由PTPN11编码,是一种去磷酸化酶,属于蛋白酪氨酸磷酸酶(PTPs)家族。SHP2作为包括Ras-Raf-MEK-ERK、JAK-STAT、PI3K-AKT-mTOR和PD-1/PD-L1通路在内的多种信号通路的汇聚节点,可以增强或拮抗与经典细胞存活相关的和具有底物特异性的免疫调节途径。
研究发现,SHP2既可以作为磷酸酶发挥显著的致癌功能,也可以以环境依赖的方式作为肿瘤抑制因子。在非活性状态下,SHP2蛋白的N-SH2结构域与PTP结构域结合并阻断其底物进入催化位点,从而导致SHP2活性受到抑制。当SH2结构域与特定的磷酸酪氨酸基序(motif)结合时,SHP2的自抑制状态被破坏,处于活化开放构象,发挥将底物去磷酸化的功能。
研究表明,SHP2参与调节生物体内多种信号通路。SHP2结合位点存在于RTK和骨架衔接子(如GAB、IRS、FRS等蛋白)中,因此这种“分子开关”确保SHP2仅在适当的细胞区域被激活。在生长因子和细胞因子信号传导中,SHP2作用于RAS的上游,并使细胞外调节蛋白激酶ERK/丝裂原活化蛋白激酶MAPK途径完全活化。
此外,SHP2的C末端酪氨酸能够响应大多数激动剂而发生磷酸化,酪氨酸磷酸化的SHP2募集接头蛋白GRB2和鸟嘌呤核苷酸交换因子SOS,有助于RAS活化。此外,SHP2通过其N-SH2结构域结合免疫检查点蛋白PD-1的磷酸酪氨酸基序(pTyr motif),参与调节T细胞的活性。因此,SHP2是一个重要的抗癌靶点。
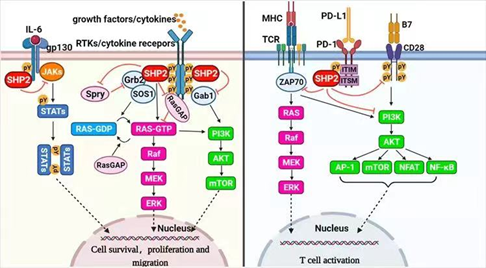
图3.SHP2介导的信号通路
3 SHP2的表达、分布及突变
SHP2是由蛋白酪氨酸磷酸酶非受体11型(PTPN11)基因编码的蛋白,这是PTPs超家族中第一个非受体原癌基因。它是一种普遍存在的蛋白质,广泛表达于多种组织类型的细胞质中,同时它也分布于细胞核和线粒体。研究发现,SHP2在多种不同的组织类型中表达,包括大脑、脂肪、甲状腺、心脏、肾上腺、肾 脏和子宫内膜等。
研究表明,转化生长因子(TGFβ)在成纤维细胞的转录和蛋白水平上均抑制了SHP2的表达。在多发性骨髓瘤中,SHP2的表达也可能通过启动子甲基化而下调。此外,一些微小RNA也可以抑制SHP2的表达。然而,目前对于SHP2蛋白的转录调控机制尚不清楚。
通常,野生型SHP2在非活性构象和活性构象之间维持动态平衡。然而,PTPN11基因的突变打破了这种平衡,并影响了SHP2磷酸酶的活性。PTPN11基因上发生两种形式的突变:功能增益(GOF)突变和功能丧失(LOF)突变(图4)。
其中,N-SH2结构域中的D61G、D61Y和E76K等GOF突变表明可以通过破坏PTP结构域自抑制状态来增强磷酸酶活性。GOF突变具有双重作用:1、通过减少N-SH2和PTP结构域之间的相互作用来增加磷酸酶活性;2、通过促进催化口袋的底物结合能力来提高磷酸酶活性。在一种独特类型的GOF突变T507K中,SHP2存在于与野生型相似的封闭自抑制构象中,但N-SH2与PTP结构域之间的相互作用较弱,这使得SHP2T507K对GAB1具有更高的亲和力。LOF突变仅发生于PTP结构域,如Y279C、T468M、Q506P和Q510E。这些突变类型可以改变催化位点,并在不影响结合能力的情况下阻断酶活性。
此外,SHP2激活的GOF突变聚集在N-SH2和PTP结构域的相互作用界面,如E76D和E76K。这些突变与一些发育障碍有关,并在白血病和多发性实体肿瘤中被发现。PTPN11的错义基因突变会导致努南综合征(Ns)和豹综合征(ls),患病率分别超过50%和80%。PTPN11的获得性GOF突变(D61Y和E76K)是最常见的突变,是幼年骨髓单核细胞白血病的主要原因。激活的SHP2突变也存在于骨髓增生异常综合征(mds,10%)、B细胞急性淋巴细胞白血病(b-all,7%)和急性髓系白血病(aml,4%)中。SHP2的高突变率导致SHP2抑制剂获得性耐药,也是困扰研发人员的一大难题。
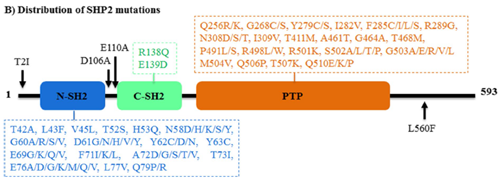
图4. SHP2的突变类型
SHP2抑制剂的最新研究进展
SHP2完成了从“不可成药”到明星靶点的角色转变。SHP2抑制剂经历了从催化位点抑制剂到变构位点抑制剂,再到SHP2降解剂的研发历程。
最早,研究人员尝试开发SHP2的催化位点抑制剂,例如含水杨酸、磺酸基团的一类化合物和某些天然产物。但是在PTP家族中磷酸酶活性域高度保守,使催化位点抑制剂在高同源性的SHP1、SHP2、PTP1B中缺乏选择性,而且结构中大多含有模拟磷酸化底物与催化中心相互作用的极性和离子功能基团存在,导致催化位点抑制剂的细胞渗透性和生物利用度较差,甚至表现出了剂量依赖的细胞毒 性、心脏和静脉毒 性等,使得这类药物难以开发成药。
2016年,诺华公司报道了第一款SHP2变构抑制剂SHP099,其以一种全新的变构抑制机制使SHP2蛋白稳定于自抑制构象。它与通过与SHP2变构口袋相互作用来稳定或抑制酶构象的变化,使活性中心不能与底物结合而发挥功能。相比于催化位点抑制剂,变构抑制剂不仅具有良好的SHP2抑制活性,比如RMC-4550、IACS-13909,对SHP2磷酸酶的IC50都在10 Nmol·L-1以内,且选择性高。此外,变构抑制剂具有成药性好、可口服利用等优点弥补了催化位点抑制剂的不足,开启了SHP2抑制剂新的研究方向。
目前,针对SHP2靶点尚无药物获批上市,多款SHP2抑制剂获批临床。其中,进展最快的处于Ⅱ期临床阶段的药物TNO-155,领跑SHP2赛道。值得注意的是,TNO155也是首 款进入临床阶段的SHP2抑制剂。SHP2作为一个明星癌症靶点,吸引了国内外多家药企布局研发。如:诺华、艾伯维、加科思、诺诚健华、苏州勤浩医药等。其中,国内进展最快的加科思的JAB-3068。据不完全统计,目前针对SHP2小分子抑制剂有:II期临床药物4个,I期临床药物11个。此外,多个研究机构布局SHP2降解剂的开发,这是未来研究SHP2的方向之一。(图5)
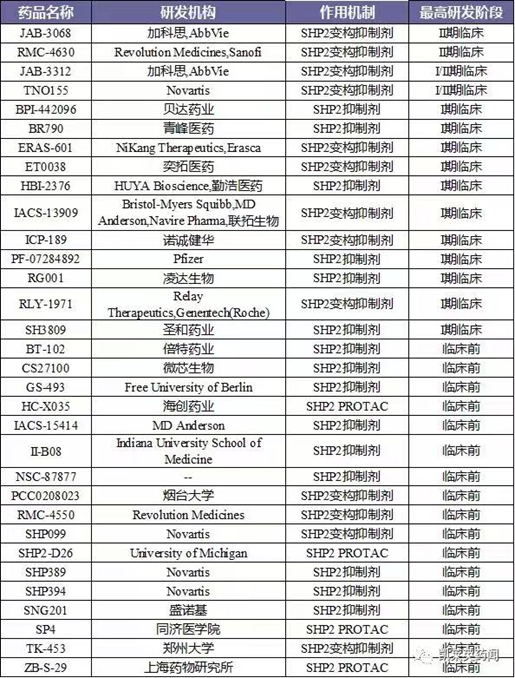
图5.SHP2抑制剂研究进展(图片来源:凯莱英药闻)
小结
过去几年里,基于SHP2抑制剂的研发已经取得了长足的进步。早期研发的靶向催化位点的SHP2抑制剂均因选择性差、生物利用度低等原因未能成药。因此,SHP2变构抑制剂的开发已成为重要的研究方向。然而,也有研究表明,目前成功开发的基于变构位点的SHP2抑制剂存在着脱靶问题。SHP2抑制剂在联合用药和替代药物的使用方面有很大的优势和空间。我们相信,SHP2抑制剂将会成为新一个药物研发的掘金之地,有望产生重磅炸 弹级别的全新抗癌药物,造福更多的患者。
参考文献:
1、凯莱英药闻.《 打破“不可成药”魔咒,SHP2抑制剂谁将率先突围?》
2、A CompreheNsive review of SHP2 aNd its role iN CaNCer. doi.org/10.1007/s13402-022-00698-1.
3、The ‘Shp’iNg News: SH2 domaiN-CoNtaiNiNg TyrosiNe phosphatases iN Cell sigNaliNg. TreNds BioChem. SCi. 28, 284–293 (2003)
4、StruCtural aNd MeChaNistiC INsights iNto LEOPARD SyNdrome-AssoCiated SHP2 MutatioNs. J. Biol. Chem. 288, 10472–10482 (2013)
5、What Have We LearNed from SH2 DomaiNs? Methods Mol. Biol. 1555, 37–43 (2017)



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























