前蛋白转换酶枯草杆菌蛋白酶/kexin 9型(PCSK9)是一种由肝 脏合成分泌的丝氨酸蛋白酶,可以与低密度脂蛋白受体(LDLR)结合,从而在细胞内吞时将其运输到溶酶体,并促进细胞内降解以防止其再循环。然而,如果循环过程中低密度脂蛋白胆固醇(LDL-C)水平异常升高,将会容易引发动脉粥样硬化性的心血管疾病(ASCVD)。因此,将PCSK9作为治疗靶点,设计出调节血浆LDL-C水平以预防ASCVD的新药是目前一种新兴的治疗方法之一。
本文主要重点介绍了基于多肽的PCSK9抑制剂的结构特点和设计、对患者的治疗作用、以及PCSK9的调节机制,以及抗PCSK9肽作为一种有希望替代单克隆抗体(MABs)来调节LDL-C水平的药物,正在成为一种有前途的新一代治疗方法。
1
背景介绍
患上动脉粥样硬化性心血管疾病(ASCVD)的患者估计每年在护理、药物和生产力损失方面的成本可能就要超过2000亿美元,同时它也是美国的头号死亡原因。高脂血症是ASCVD的一个公认的风险因素,包括动脉粥样硬化、中风、心力衰竭、心肌梗死(MI)和冠心病(CHD)。
与健康人相比,高胆固醇[低密度脂蛋白胆甾醇(LDL-C)升高]的人患心脏病的风险增加2倍。美国高LDL-C成年人的人数为7370万,即31.7%。在接受治疗(他汀单独或联合依折麦布)的约48.1%的高LDL-C患者中,只有29.5%的受试者病情得到控制。家族性高胆固醇血症(FH)是导致LDL-C终身升高和冠心病风险显著增高的常见形式。
前蛋白转换酶枯草杆菌蛋白酶/kexin 9型(proprotein convertase subtilisin/kexin-type 9,PCSK9)被发现为FH的遗传决定因素,近年来成为降低高胆固醇血症患者LDL-C水平的潜在靶点。PCSK9通过降解低密度脂蛋白受体(LDLR)来调节血清中LDL-C水平。因此,PCSK9抑制剂已成为一种血脂控制新疗法,可减轻动脉粥样硬化的发病机制,降低心血管事件和死亡率。
PCSK9在血管壁、肠、肾和脑中表达,在这些组织中,除了提高脂质外,它还参与了其它的功能,尽管有新的证据表明PCSK9可以发挥多种多效性作用,但它的主要功能仍然是控制胆固醇的稳态。(图1)
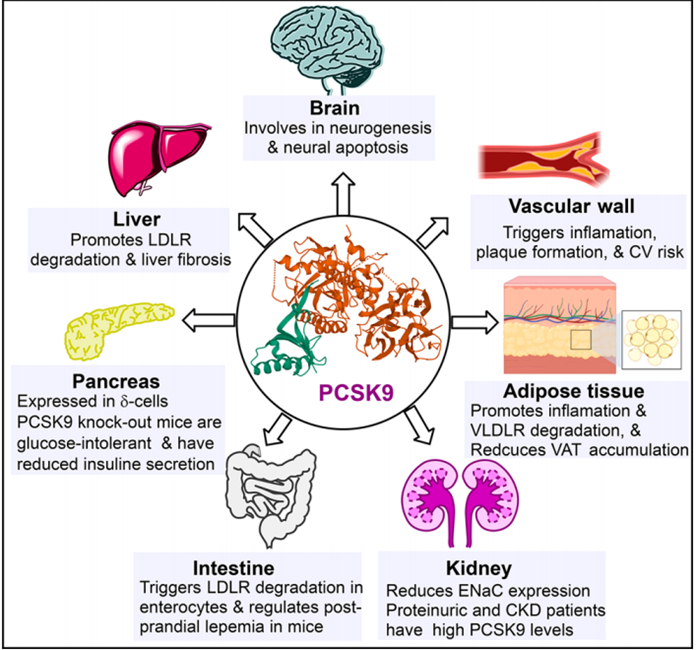
图1. PCSK9发挥多效性效应的示意图。
2
高胆固醇血症的治疗
许多降脂疗法已显示出治疗高脂血症的积极结果。其中以羟甲基戊二酰辅酶A还原酶(HMG-CoAR)抑制剂(他汀类药物)、胆固醇酯转移蛋白抑制剂(anacetrapib)、胆固醇吸收抑制剂(依折麦布)、和PCSK9抑制剂等为代表。
如果饮食和生活方式的改善未能降低LDL-C水平,通常通过他汀类药物作为一线药物治疗。然而,他汀类药物治疗与不良副作用相关,包括他汀类药物相关肌肉症状、肝毒 性、发生2型糖尿病的可能性和出血性卒中风险。此外,约50%使用他汀类药物的个体无法将其LDL-C水平降低至预期水平,从而使患者发生脑血管疾病的风险升高。
此外,他汀类药物治疗可上调PCSK9,进一步降低他汀类药物的有效性。依折麦布是降低LDL-C的二线药物,可阻止肠道吸收胆固醇。对于接受最高剂量他汀类药物治疗但仍需要额外降低LDL-C的个体或他汀类药物不耐受的患者,则需要依折麦布作为辅助治疗。在LDL-C和FH水平最高的个体中,高强度他汀类药物单药治疗或与依折麦布联合治疗不足以达到预期的LDL-C水平。
而PCSK9抑制剂比他汀类药物的优势是,他汀类药物不会显著降低脂蛋白(a) [LP(a)]水平,这是脑血管疾病(CVD)的一个独立危险因素,而PCSK9抑制剂则会降低。这是因为PCSK9参与LDL相关蛋白-1的降解,该蛋白分解LP(a),因此,抑制PCSK9会提高LDL相关蛋白-1的水平,从而增加LP(a)的分解。在表1中对比了他汀类药物和PSCK9抑制剂的临床治疗影响。

表1. 高强度他汀类药物和PCSK9抑制剂对临床血浆参数影响的观察
3
PCSK9
3.1 PCSK9的结构与生物学功能
PCSK9是丝氨酸蛋白酶家族的第九个也是最后一个成员,该家族包括原蛋白转化酶1(PC1),PC2,PC4,PC5,PC7,Furin,配对碱性氨基酸裂解酶4(PACE4),枯草杆菌蛋白酶Kexin同工酶1和PCSK9.这个丝氨酸蛋白酶家族更类似于细菌枯草杆菌蛋白酶。前八个转化酶(PC1到SKI-1)裂解分泌性蛋白前体以产生调节发育和代谢的生物活性肽、多肽和激素。
然而,PCSK9自身裂解,不再作为蛋白酶发挥作用。相反,它起非酶作用,并增强LDLR的内体和溶酶体降解(在LDL-C稳态中至关重要)。人PCSK9(hPCSK9)基因编码72kDa的酶原,含692个氨基酸,由12个外显子和11个内含子组成。11个PCSK9具有信号肽、原结构域、与枯草杆菌蛋白酶相似的催化结构域(CD)和丝氨酸蛋白酶中具有独特折叠的可变C端结构域。
PCSK9蛋白的晶体结构如图2A(PDB:2PMW)所示。控制PCSK9细胞定位的基序可能存在于羧基末端结构域(残基452至692)中,其特征是富含半胱氨酸和组氨酸的区域(CHRD)。该结构域与抵抗素同型三聚体具有结构相似性,抵抗素同型三聚体是一种与糖尿病和肥胖有关的微小细胞因子,对PCSK9-LDLR复合体的内溶酶体降解至关重要。产生的PropCSK9在VFAQ152SIP位点经历了自催化裂解,形成了一个13 kDa的前结构域和一个成熟的PCSK9结构域(图2B),为了使PCSK9在生物学上运作,需要原结构域和PCSK9的异源二聚体,但不需要PCSK9的酶活性为了防止催化三联体排列和保护邻近的蛋白质免受其丝氨酸蛋白酶活性的影响,PCSK9的蛋白酶活性被抑制。成熟的PCSK9在肝细胞外以一种完整的异源二聚体(62+13kDa)的形式分泌。PCSK9已被证明在控制胆固醇稳态方面具有重要功能。
PCSK9最显著的特征是它与LDLR的EGF-A结构域相互作用,并通过将其引导到溶酶体来促进受体降解。PCSK9:EGF-A复合物的晶体结构如(图2C)所示(PDB:3bps)。值得注意的是,与PCSK9的起源无关,LDLR调节其从等离子体中的清除。LDLR当附着在LDL-C上时,进入细胞,然后在内体中与LDL-C解离,如果不与PCSK9偶联,最终会穿梭到细胞表面。
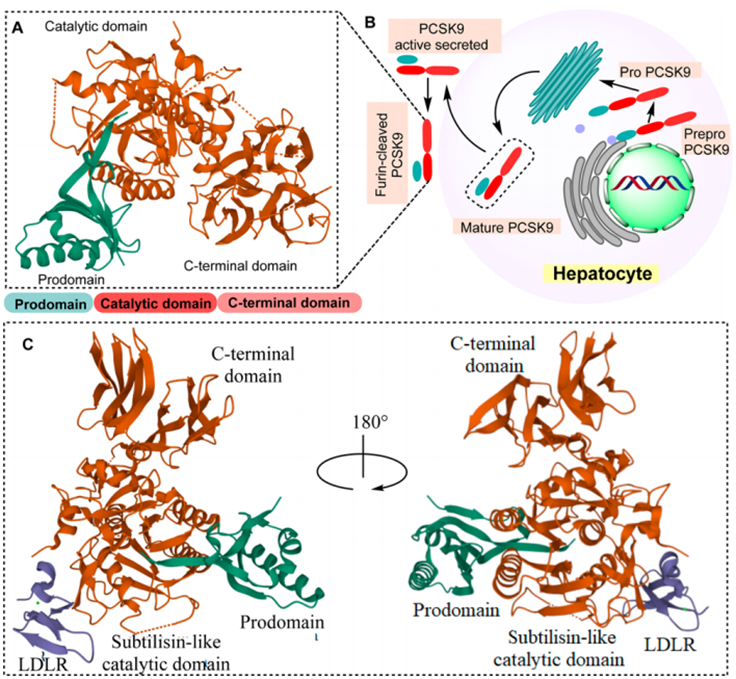
图2. (A) PCSK9蛋白的晶体结构显示了多种结构域,包括原结构域(绿色)、C端结构域(棕色)和类似枯草杆菌蛋白酶的催化结构域(PDB:2PMW)。(B) PCSK9在肝细胞中成熟过程示意图。(C) PCSK9:EGF-A复合体的晶体结构表明了EGF-A结构域的fldlr(蓝色)和PCSK9的不同结构域(PDB: 3BPS)。
3.2 PCSK9在动脉粥样硬化形成中的作用
动脉粥样硬化斑块是一种脂蛋白驱动的疾病,其特征是动脉壁内膜增生、炎症、坏死和钙化。肝、肾和小肠是循环PCSK9的主要来源。PCSK9也由内皮细胞、血管平滑肌细胞(VSMC)合成,并在较小程度上由巨噬细胞合成。这些观察表明,用PCSK9刺激的巨噬细胞可能抑制泡沫细胞的形成,从而形成动脉粥样硬化。结合这些发现将PCSK9与斑块的形成联系起来。随着巨噬细胞从单核细胞(几乎无法检测到)到分化单核细胞(在那里高表达)再到完全分化的巨噬细胞,PCSK9基因在巨噬细胞中的表达逐渐增加。有趣的是,用Evolocumab抑制PCSK9改善了HIV+血脂异常患者的冠状动脉内皮功能。当HPCSK9通过增强骨髓来源的LY6ChIgH单核细胞的病变浸润而改变斑块结构,进而导致它们以LDLR依赖的方式分化为巨噬细胞时,PCSK9可能促进动脉粥样硬化形成中的炎症的想法首次被实现。
这些结果与HPCSK9表达增加脂多糖刺激的小鼠腹腔巨噬细胞肿瘤坏死因子-α(TNF-α)和白细胞介素-1β(IL-1β)mRNA表达的事实一致,这一事实后来在原代人巨噬细胞中得到证实。
PCSK9的缺失导致了一种通过LDLR以外的途径易感动脉粥样硬化的小鼠品系中致动脉粥样硬化脂蛋白较少的表型。LDLR、APOBEC1和PCSK9缺失小鼠的脂蛋白暴露于内皮细胞后,内皮细胞产生的ICAM-1、IL-1β和趋化因子(包括CCL2和CCL-7)减少,所有这些都促进了单核细胞向血管壁的粘附和浸润。在参与动脉粥样硬化炎症负担的众多炎症因子和清道夫受体中,凝集素氧化脂蛋白-1(LOX1)起主导作用。
LOX-1与PCSK9呈正相关,LOX-1促进PCSK9,PCSK9促进LOX-1和氧化LDL的吸收。此外,小鼠重组PCSK9可促进TNF-α诱导的巨噬细胞中清除受体(如CD36和LOX-1)的表达和氧化LDL的吸收。LDLR相关家族,如LRP5,也有助于脂质内化到巨噬细胞中。尽管PCSK9和LRP5不会改变彼此的表达,但它们在核周区域的共定位允许它们直接相互作用。免疫沉淀研究表明,聚集的LDL刺激促进脂质负载巨噬细胞的形成。
如前所述,树突状细胞或巨噬细胞中脂蛋白产生的脂质的积累激活了一种称为炎症体的多蛋白复合物。人类动脉粥样硬化病变具有三个结构域的增加表达,这三个结构域组成核苷酸寡聚结构域(NOD)、富含亮氨酸重复(LRR)和NLR家族含pyrin结构域(NLRP3)炎症体:末端pyrin结构域、中央核苷酸结构域(NACHT结构域)和C末端富含赖氨酸结构域。NLRP3复合物的形成导致自催化Caspase 1激活。在炎症和PCSK9之间的纠缠相互作用中,γ干扰素促进mRNA表达,而NLRP3途径调节PCSK9分泌。在不产生LDLR基因敲除的情况下,研究转基因或敲除小鼠的动脉粥样硬化发病机制的体内实验进一步支持了PCSK9在动脉粥样硬化过程中的关键参与。
3.3抗PCSK9的药物类型
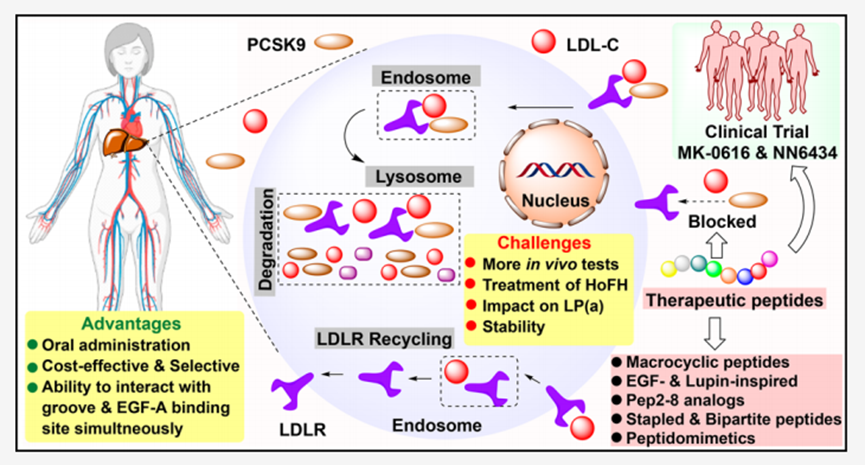
PCSK9抑制剂是利用针对PCSK9生产、加工和结合的策略开发的,以控制循环LDL-C水平(图3)。开发小分子的常规方法尚未对 PCSK9 有效。这同样适用于开发基于蛋白质的疫 苗接种,这些疫 苗可引导免疫系统产生抗 PCSK9 抗体以中和循环中的 PCSK9。然而这些疫 苗也未达到标准预期。因此,探索 CRISPR-Cas9、小干扰核糖核酸 (siRNA)、反义寡核苷酸 (ASO)、单克隆抗体、adnectin、anticalin 和肽等新药模式的研究势头强劲。
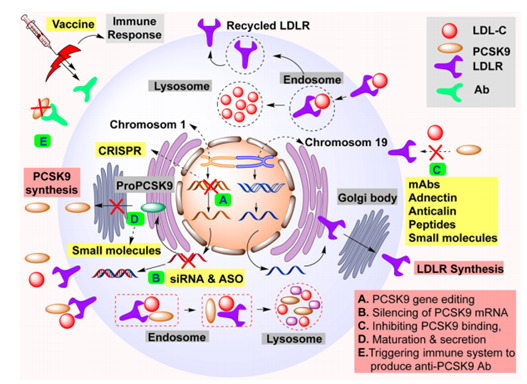
图3. 针对 PCSK9 生产、加工和与 LDRL 结合以管理 LDL-C 水平的各种药物类型。
3.3.1抗PCSK9的小分子治疗。
使用小分子靶向PCSK9有几个优点,包括更好的稳定性、更低的生产成本和更简单的管理(表2,条目1)。这些小分子通过阻断PCSK9:LDLR PPIs,干扰PCSK9的成熟和分泌,或抑制PCSK9的表达来降低LDL-C。然而,小分子(质量500 DA)不能有效抑制PCSK9:EGF-a PPIs,因为PCSK9的表面相对平坦、无特征且不可成药。上述假说从小分子介导的PCSK9抑制只产生一个足够感兴趣的潜在候选药物,即CVI-LM001(1)这一事实中得到了明显的证明。CVI-LM001(1)是紫堇碱的氟苯磺酸衍生物,通过抑制PCSK9基因的表达而起作用(图4)。在IB期研究(CTR20160744)中,300毫克剂量的CVI-LM001(1)使LDL-C水平升高的参与者的LDL-C水平降低26.3%,TC降低20.1%,载脂蛋白降低17.4%。

图4. 临床小分子抗 PCSK9 疗法 1 和 2 的化学结构。

表2. PCSK9抑制药物类型的比较
3.3.2 Adnectin 作为 PCSK9 靶向治疗药物
Adnectin是为延长给药间隔而开发的新模式。Adnectins是重组融合蛋白,与MABs相比具有许多优点,如体积小、费用低、生产容易、亲和力高、起效快(表2,条目7)。在I期临床研究中,BMS-962476是一种与聚乙烯偶联的多肽,使LDL-C水平下降48%,使游离PCSK9下降>90%(NCT01587365)。PCSK9和Adnectin的X射线共晶结构很好的揭示了对PCSK9的抑制机制(BMS-962476的祖细胞)。共晶的每个晶胞包含两个PCSK9-肾上腺素复合物,如图6A(PDB ID:4OV6)所示。LDLR与PCSK9-Adnectin结构的重叠表明Adnectin 1459D05位于LDL受体结合表面附近,以阻止PCSK9:LDLR PPIs发挥作用(图5B)。
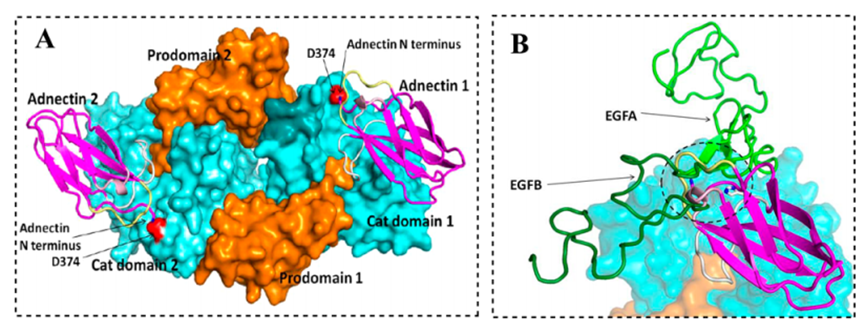
图5. (A)PCSK9与Adnectin 1459D05的共晶结构显示一个包含两个PCSK9-Adnectin复合物的晶胞(PDB ID:4OV6)。(B)LDLR(绿色)与PCSK9/Adnectin结构的重叠,表明LDLR与Adnectin之间的空间冲突(PDB ID:3P5C)。
3.3.3 作为PCSK9靶向治疗药物的多肽
与其他抑制PCSK9的策略相比,多肽有几个好处(表2,条目9)。例如,小分子由于体积小而缺乏选择性,不能有效地与PCSK9等不可药物作用的靶点结合。类似地,大型生物制剂,如MABS,价格昂贵,不稳定,缺乏药物性质。相反,多肽治疗有一个理想的大小(500-5000Da),这使得很容易使它们功能化以增强它们的药物样特征,如生物利用度、稳定性、亲和力和特异性。进一步多肽模拟复杂蛋白质的结构,从而允许它们与PCSK9相对平坦和大的表面相互作用以破坏PCSK9:LDLR PPIs。
此外,多肽治疗药物,像小分子药物一样,比大型生物药物更容易制造,这降低了生产成本。因此,多肽在许多方面占据了小分子和生物药物之间的独特位置,并被认为是可能干扰PCSK9功能的重要药物先导物(图6)。

图6. 使用 BioRender.com 创建的大型生物和小分子疗法与肽模式的比较。
4
基于PCSK9多肽抑制剂的临床研究
两种肽 PCSK9 抑制剂 MK-0616 (5) 和 NNC03850434 (NN6434, 6) 已进入临床试验阶段。大环肽以其对蛋白水解降解的高稳定性而闻名,并具有破坏大而扁平目标的 PPI 的固有特性。考虑到 PCSK9 的 PPI 与 LDLR 的相似特征,LDLR 是一种环肽抑制剂PCSK9(3)利用mRNA展示技术被设计(图7)。然后将HIT肽3优化为一种高效的、代谢稳定的PCSK9抑制剂,其分子量降低为4。然后将第二代肽4精制成三环的、口服生物可用的MK-0616(5)。MK-0616(5)是一种新型大环化合物,其亲水性、抑菌能力(约10万×)和体内活性均优于初始先导化合物。在I期临床研究中,单剂量10-300毫克的MK-0616(5)耐受性良好,并使健康参与者血清中PCSK9水平下降90%。
另一种治疗高胆固醇血症的基于肽的长效PCSK9抑制剂是诺和诺德的NN6434(6)。目前正在对NN6434进行多次第一和第二阶段临床研究。2022年,启动了一项I期试验,以测试三种不同的NN6434(6)片剂配方来治疗高胆固醇血症(NCT05333107)。在II期临床试验中,他们还计划评估15毫克NN6434(6)对CVD患者的有效性(NCT04992065)。
图片
图7. (A)Merck大环MK-0616(5)的设计和生物活性。(B)与PCSK9上LDLR结合位点结合的4的晶体结构(PDB ID:6XIE)。
5
总结与展望
长期以来由于PCSK9的表面相对平坦且无特征,因此开发能够阻止PCSK9与LDLR相互作用的小分子非常具有挑战性。幸运的是,多肽作为一种特殊的化学类型,为大生物制剂和小分子提供了一种有希望的替代方案,并成为抑制PCSK9的有吸引力的下一代治疗药物。
为了提高PCSK9抑制剂的刚性、蛋白水解稳定性、细胞通透性和口服生物利用度,许多制药行业都在广泛地研究肽的环化。例如,最 先进和最有效的抗PCSK9肽是大环,如诺华的13PCSK9I和默克的MK-0616。制药行业对开发大环肽作为PCSK9拮抗剂的兴趣和投入加大,进一步加强了这种方法治疗高胆固醇血症的治疗前景。因此,我们非常期待发展更多安全有效的PCSK9抑制剂来造福患者。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























