介绍
具有亲电性的小分子药物可以与内源性靶蛋白的亲核氨基酸残基形成共价键,不可逆地改变目标蛋白的功能。由于与蛋白的结合时间增长,化合物可以持续增强药理作用,实现异构体的选择性抑制,克服肿瘤细胞的获得性耐药。
为了减小共价小分子脱靶引发毒性的风险,共价药物的研究集中于开发靶向共价抑制剂(TCI),其定义为:通过合理设计,将亲电性的,具有共价反应活性的靶头引入靶蛋白的可逆配体上,使其能够选择性地共价结合到靶蛋白质上持续发挥抑制作用。
为了达到较高的选择性,TCI的靶头活性需要经过仔细调整,使其在生理条件下保持稳定,但活性又足够让其共价结合到靶蛋白。除了反应活性,靶头的化学选择性、反应方式、分子大小以及共价的可逆性也会影响TCI的最终抑制效果。
在本篇综述中,我们将概述靶向蛋白质共价修饰这一化学手段的研究进展及其在TCI设计中的应用。尽管文中介绍的靶头不是所有的都被应用到了体内研究,但考虑其独特的作用机制与反应模式,其仍有很大可能在未来的临床研究中大放异彩。
Michael受体以及烯类和炔类亲电靶头
α,β-不饱和酰胺类Michael受体是在TCI中应用最为广泛的靶头。以此为靶头靶向酪氨酸激酶如表皮生长因子受体(EGFR)和Bruton’s酪氨酸激酶(BTK)的TCI目前已有8个被FDA批准上市用以治疗癌症(图1)。作为这些TCI药物的靶头,丙烯酰胺或2-丁炔酰胺通过与目标激酶ATP结合位点半胱氨酸的巯基共价结合使靶蛋白不可逆地失活。
基于活性的蛋白质谱学研究(ABPP)也进一步佐证了其共价结合模式。由于缺乏明确的,能容纳小分子药物的结合口袋,G12C突变型KRAS蛋白长期以来被认为“无药可靶”。直到以α,β-不饱和酰胺为靶头的TCI药物出现才打破了这一僵局。Sotorasib (AMG-510) 现已被FDA批准用于治疗 KRAS G12C突变的非小细胞肺癌。Adagrasib (MRTX-849)已与2022年被FDA批准治疗KRAS G12C突变的非小细胞肺癌和胰腺癌(图1B)。
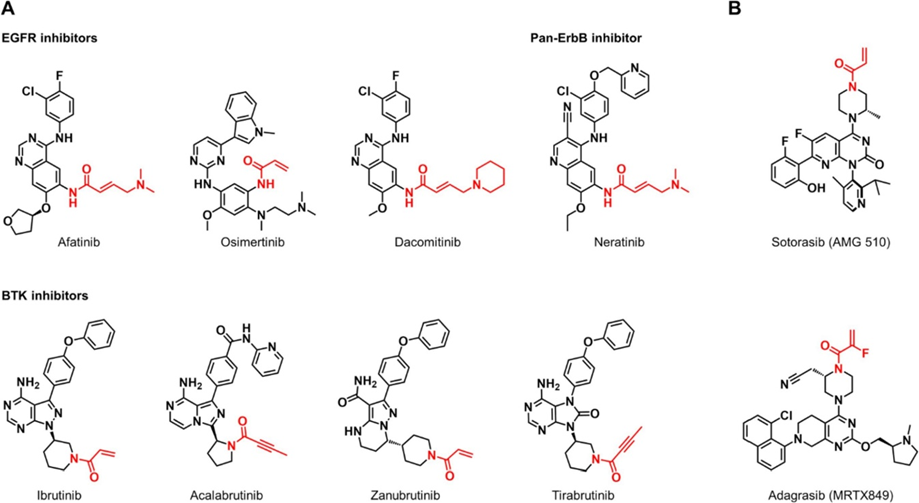
丙烯酰胺的一个显着特征是其能够自由调整对硫醇的反应活性,具体取决于烯烃位置的取代基。β位的氨甲基是靶向激酶TCI中的常见结构。虽然丙烯酰胺的β-甲基化会减慢其与硫醇的加成速度,但β-氨甲基丙烯酰胺则表现出与未取代的丙烯酰胺相当的硫醇反应性。
实验和DFT研究表明,氨甲基取代基在质子化状态下作为吸电子基团以增强α,β-不饱和酰胺的亲电反应性。而Gao课题组最 先发现,在α位引入一个氟原子会显着降低丙烯酰胺的反应性。α-氟取代的丙烯酰胺被用作KRAS G12C抑制剂阿达格拉西的靶头,以延缓体内GST介导的谷胱甘肽(GSH)结合型药物代谢物的产生。
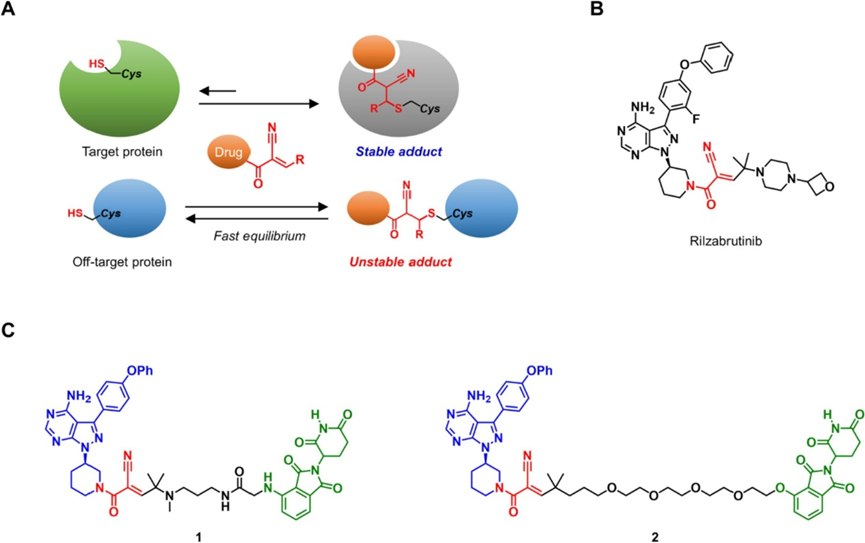
与之相对的是,在α位引入吸电子氰基则会同时促进硫迈克尔加成以及逆迈克尔反应的进行。这使得半胱氨酸的巯基对双重活化的氰基丙烯酰胺的亲核加成成为了完全可逆的过程(图2A)。
汤顿课题组就以此设计了靶向p90核糖体S6激酶RSK和BTK的可逆共价抑制剂。他们发现,TCI与目标蛋白的共价加合物除了依赖可逆的共价键,也会通过非共价相互作用得到稳定,而由于TCI无法与目标蛋白以外的形成有效的非共价相互作用,脱靶的硫醇加合物最终将通过反向逆迈克尔反应轻松消除。这种策略可以最大限度地降低共价药物在体内非特异性结合引发毒性的风险。尽管可逆共价抑制剂在靶点的停留时间可能比不可逆结合剂短,但他们发现基于氰基丙烯酰胺的BTK抑制剂的保留时间可通过改变β-侧取代基的大小来调节,并在对大鼠进行的口服给药试验中得到证明。
Rilzabrutinib(PRN1008)已获得快速通道指定,并且正在进行治疗免疫性血小板减少症的3期研究(图2B)。近年来,蛋白质水解靶向嵌合体(PROTAC)等化学蛋白质降解剂已成为抑制蛋白质活性的强力策略。已有报导文献利用基于丙烯酰胺的共价抑制剂作为PROTAC的靶蛋白结合配体。还研究了基于氰基丙烯酰胺的BTK共价可逆抑制剂在PROTAC设计中的应用(图2C)。理论上讲,可逆的共价PROTAC可以实现目标特异性的提高和目标蛋白的催化型降解。
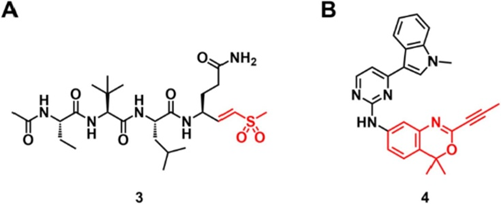
丙烯酰胺型迈克尔受体通常对整个蛋白质组中的半胱氨酸残基具有化学选择性。除丙烯酰胺以外,其它类别的迈克尔受体也被积极运用到TCI的研究中。乙烯基砜可与硫醇发生迈克尔加成反应,通常用于病毒和寄生虫半胱氨酸蛋白酶的不可逆抑制,也包括严重急性呼吸系统综合症冠状病毒2 (SARS-CoV-2)的主蛋白酶(图3A)。然而,基于乙烯基砜的共价抑制剂很少在体内环境中进行测试,这可能是因为它们具有高内在反应性。最近,阿斯利康的团队也报道了用炔基苯并恶嗪和二氢喹唑啉这两种亲电性靶头作为丙烯酰胺的替代品。
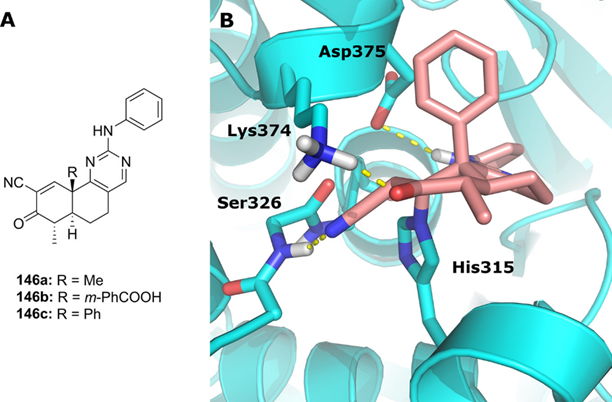
在诸多Michael亲电性靶头中也有报道能够共价连接到靶蛋白的赖氨酸或组氨酸残基上。组氨酸的咪唑基团也是一个非常强效的亲核基团,可以用来被共价修饰。Jakob课题组首次运用α-氰基酮结构靶头实现了对位于野生型异柠檬酸脱氢酶(IDH)1的NADPH结合口袋中His315的共价修饰。他们进一步的实验结果表明化合物146C只会选择性地与His315形成可逆的氮杂迈克尔加成物,而化合物中的α-氰基则会在结合口袋处形成关键的氢键引导TCI对靶蛋白的识别(图4)。赖氨酸的胺基残基虽然能够被迈克尔受体所修饰。但大多数Michael受体对“较软”的半胱氨酸硫醇更具反应性,所以靶向赖氨酸ε-氨基可能需要活性更高的亲电受体。但后者有可能表现出半胱氨酸介导的脱靶效应
Michael受体是TCI设计中应用最为广泛的亲电性靶头,主要用于靶向半胱氨酸,通过在其α位引入氰基可以加快Micheal反应的双向进行,可用于制作可逆型共价靶头,已经有多篇综述报导通过在α和β位引入取代基来达到延长可逆TCI在靶点的保留时间,优化靶头自身的反应性和化学选择性甚至引导TCI选择性地与其他亲核性氨基酸残基诸如组氨酸实现共价修饰。考虑到赖氨酸较弱的亲核性,可以选择磺酰胺类Michael受体为靶头,但可能会出现对半胱氨酸的脱靶效应,所以Michael受体不作为赖氨酸修饰的首选靶头。
α-卤代酰胺类亲电靶头
α-卤代乙酰胺是另一类常见的靶向半胱氨酸亲电靶头,它通过SN2机制与硫醇反应,其反应速率取决于亲核试剂和离去基团的活性。α-氯乙酰胺在生理水性条件下非常稳定但其活性又足够共价修饰。α-卤代酰胺的α位亚甲基由于离去基团的和羰基的吸电子作用下有更强的亲电性,并且这两个取代基也有助于其SN2-过渡态的稳定。如果+M取代基与羰基相连,那么α-卤代甲基羰基化合物的反应性就会降低。因此,α-卤代酯的反应性低于相应的酮,并且相应的酰胺反应性更低。
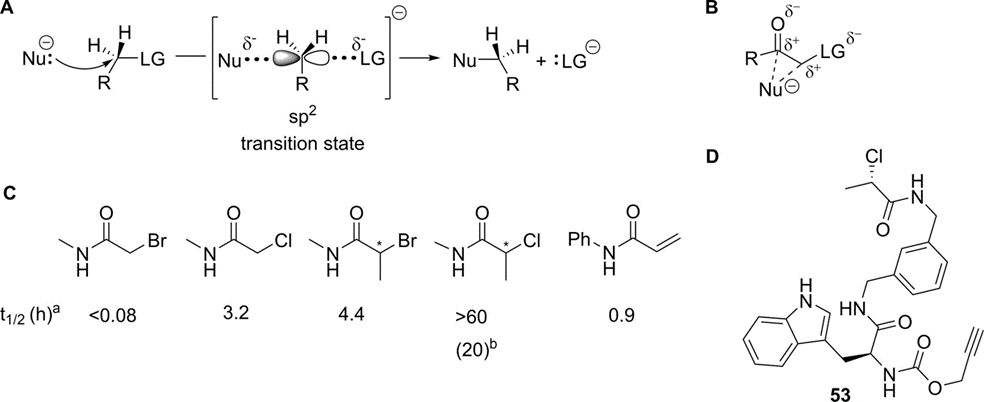
另一方面,α-卤代酮的反应性会随着卤素离去能力的增加而增加(I > Br > Cl > F)。在这其中,α-卤代乙酰胺用途广泛且可用于标记各种亲核试剂(通常是Cys,但也包括Lys、His、Tyr,活化的Ser和Thr等)。通过在反应位点附近引入空间位阻,进一步降低α-卤代乙酰胺的反应性。因此α-溴丙酰胺的反应速率低于α-氯乙酰胺的反应速率(图7C)。但在具体情况下,我们可能需要反应活性低的靶头来提高整体化合物的选择性。Allimuthu课题组正是利用了α-氯丙酰胺的低反应性和特异反应性,合成了(S)-CW3554(图5D)作为蛋白质二硫化物异构酶A1 (PDIA1)的不可逆抑制剂,其在HEK293细胞中具有良好的选择性。
总体而言,对于α-卤代酮类这一类靶头,最为常见的是α-卤代酰胺,其适合用来靶向非催化位点的半胱氨酸。该类靶头的反应活性与α位的离去基团离去能力和空间位阻相关。将α位的卤素替换成别的离去基团,或者在α位引入大位阻的取代基都将影响靶头的活性以及选择性。
羰基衍生物
4.1酯、酰胺、胺基甲酸酯以及脲
通过活化酯和酰胺对具有催化活性的氨基酸残基进行酰基化修饰,如阿司匹林和β-内酰胺类抗生素等经典共价药物。此类靶头使用了各种酰化剂,包括酯、酰胺、氨基甲酸酯和脲。通常,这些靶头优选与具有亲核性的胺基(赖氨酸和蛋白质游离N-末端)或活化的羟基(丝氨酸、苏氨酸和酪氨酸)反应,从而与靶向半胱氨酸的共价抑制剂有所区分。N-羟基琥珀酰亚胺基和多氟苯基酯等活化酯对赖氨酸残基表现出广谱反应活性。然而,这些活化酯的反应性太强反而降低了自身的化学选择性,并且也会对体内各类水解酶敏感,加大了体内应用的难度。

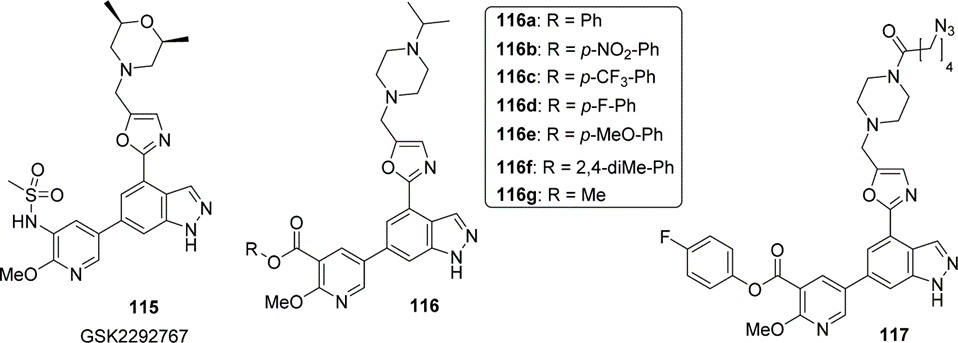
Campos团队最近运用活性酯靶向赖氨酸侧链的能力,合成一种有强效的靶向PI3Kδ的共价抑制剂,由于临床候选药GSK229276 115已被证实是依赖磺胺部分来与保守的Lys779 ε-氨基相互作用,他们就将原磺胺结构替换成了活性更强的活化酯 (116a-f),并且最终优化出了对氟苯酚甲酸酯这一新靶头,与之对应的化合物117展现出对PI3Kδ更强的抑制作用以及更好的化学选择性(图7)。而活化的胺基甲酸脂则被用于修饰有催化活性的丝氨酸。
4.2醛、酮和乙腈
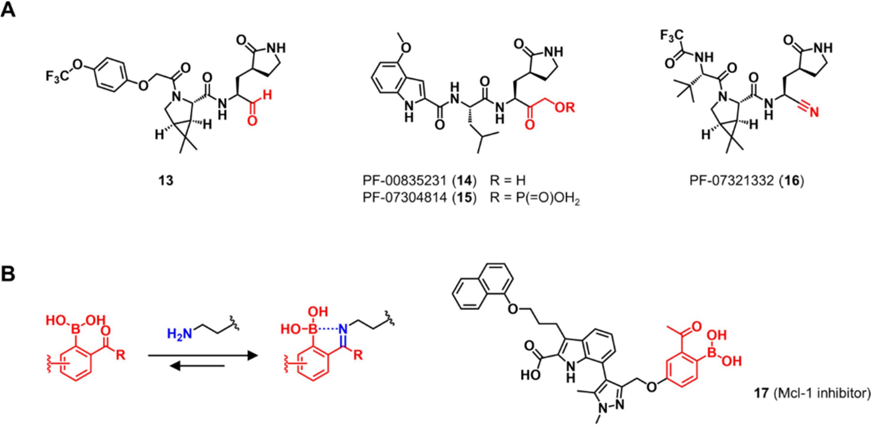
醛和酮对亲核性氨基酸残基表现出广谱的反应活性。醛和酮聚合的肽类似物通过与羟基和巯基形成半缩醛复合物来达到可逆的共价效果。例如,α-酮酰胺化合物telaprevir和boceprevir与丙型肝炎病毒NS3/4A的具有催化活性的丝氨酸残基发生反应。腈也可用作靶向蛋白酶的可逆共价靶头。维格列汀和沙格列汀是二肽基肽酶4 (DPP-4)抑制剂,用于治疗2型糖尿病。它们的腈部分与DPP-4的催化活性丝氨酸残基形成可逆的共价键。
2019年新型冠状病毒(COVID19) 的全球大流行促使针对其致病病原体SARS-CoV-2的抗病毒药物的开发。上述提及的醛、酮和乙腈这几个可逆靶头已被用于开发靶向SARS-CoV-2 Mpro的共价抑制剂,其中一些已被证明在体内有效(图8A)。醛酮类靶头也可以与分子中的硼酸基团联合,稳定抑制剂与靶蛋白的结合(图8B)。与芳香醛或酮相邻的硼酸不仅加速亚胺与赖氨酸氨基的形成,而且稳定形成的亚胺加合物。Su课题组利用这种化学方法开发了靶向髓细胞白血病1 (Mcl-1)的可逆共价抑制剂。
氟化磺酰类靶头
氟磺酰是共价修饰赖氨酸ε-氨基的最适合的靶头之一,其还被用于靶向酪氨酸、活化的丝氨酸或苏氨酸残基,并且对半胱氨酸和组氨酸侧链也具有反应活性。在这类结构中,氟磺酰亚胺拥有出色的稳定性、多功能性和可调节性,是很好的酪氨酸和赖氨酸靶头。芳基氟硫酸盐在pH=7.5时非常稳定,既不会发生水解也不会被N-α-乙酰基赖氨酸或酪氨酸取代,由于其反应性较低一般作为潜在的亲电靶头,只有在特定条件下,比如结合到特定蛋白位点后,结合口袋区的氨基酸残基能够提供氢键供体或者亲电基团来稳定可离去的氟离子,这时候芳基氟硫酸盐的活性才能被唤醒。
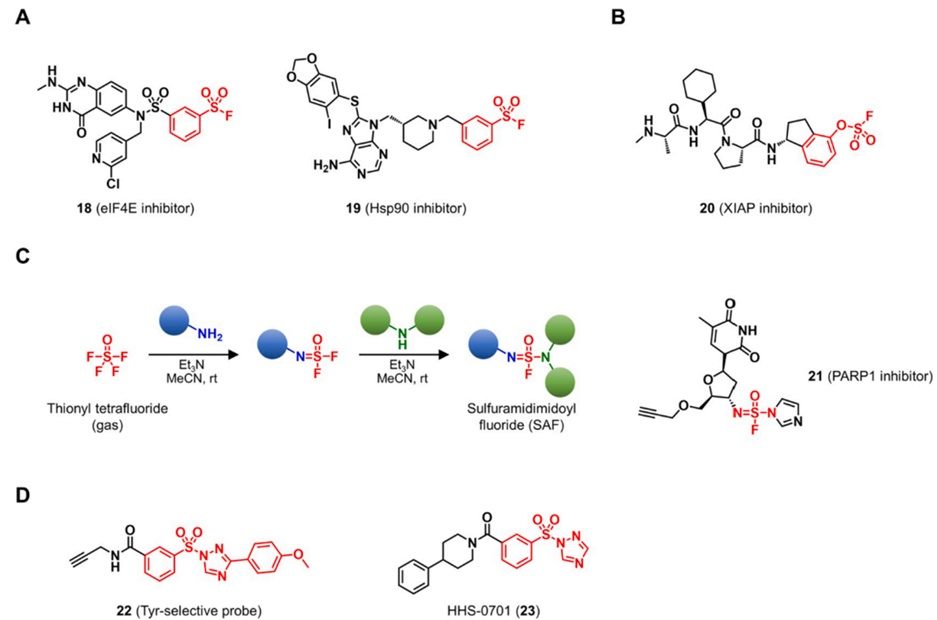
Taunton课题组最近报道了靶向真核翻译起始因子4E (eIF4E)和Hsp90的赖氨酸共价抑制剂(图9A)。Pellecchia课题组报导了X连锁凋亡抑制蛋白(XIAP)的共价配体和靶向Mcl-1的共价肽。这些基于芳基氟磺酰的药物可以通过靶向蛋白质表面的赖氨酸来抑制PPI。Pellecchia课题组报导了一种基于芳基氟硫酸盐的共价XIAP抑制剂,其靶向BIR3结构域的Lys311(图9B)。他们的芳基氟硫酸盐作为潜在的亲电子靶头,仅在蛋白质靶标的结合口袋中产生反应活性。
Kelly和Sharpless小组以亚硫酰四氟化物气体模块化合成亚硫酰亚胺基氟化物SAF(图9C)。其合成的SAF 21靶向到NAD+结合位点的Tyr907并不可逆地抑制其在活细胞中的活性。Hsu团队将苯基氟磺酰的离去基团由原本的氟更改为三唑,用于靶向活细胞中的酪氨酸和赖氨酸残基。有趣的是,对三唑离去基团的化学修饰提高了靶头对酪氨酸的化学选择性。与氟磺酰对应体相比,苯基磺酰三唑与蛋白质的反应性显着提高。他们合成的HHS-0701 (23)被鉴定为前列腺素还原酶2 (PTGR2)的共价抑制剂。
其他类型靶头
6.1芳卤和芳基磺酮
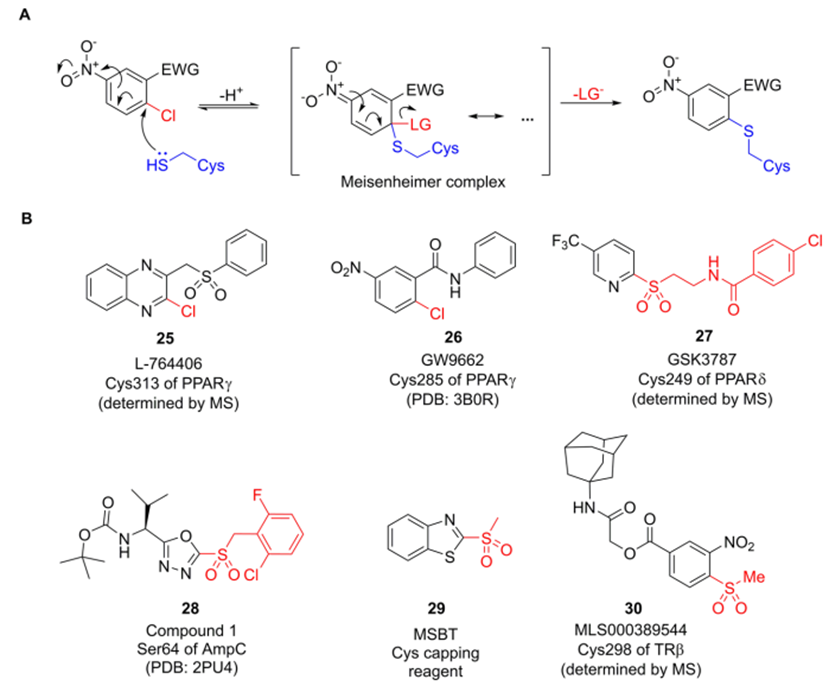
亲核芳香取代(SNAr)反应在蛋白质靶向共价修饰领域有着悠久的历史。通过改变芳环取代基和离去基团的电性,我们可以轻松地调整SNAr亲电子试剂的反应性。4-氯硝基苯在蛋白质组学中表现出对半胱氨酸的化学选择性,而二氯三嗪对赖氨酸具有高度选择性。以此类结构为靶头的药物,例如2-氯喹喔啉L-764406(25,图10B),2-氯苯甲酰胺GW9662 (26)和2-磺酰基吡啶GSK3787(27)被用于靶向过氧化物酶增值因子激活受体(PPAR)的非催化活性半胱氨酸。2-磺酰恶二唑,如28所示,通过与催化丝氨酸共价结合以阻断AmpC β-内酰胺酶,或作为马来酰亚胺替代物用于促进血清稳定抗体偶联物的产生。
6.2应变分子的亲电试剂
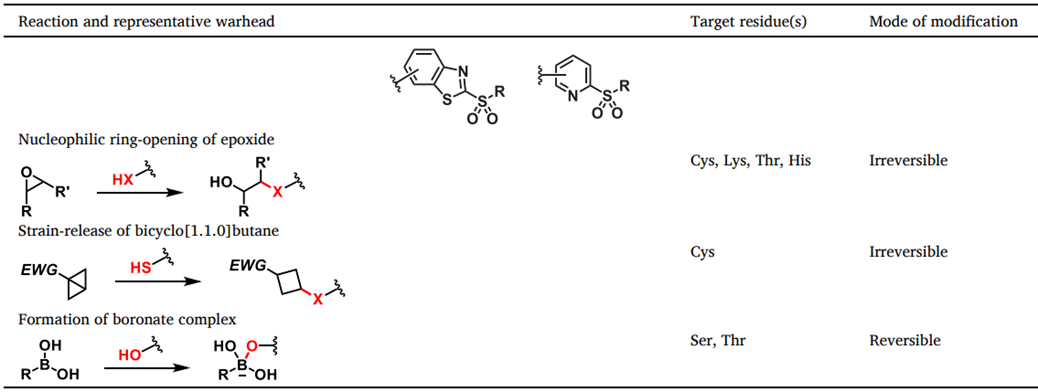
应变分子的独特反应性已在共价药物研究中成功得到利用。环氧化物存在于各种具有生物活性的天然产物和共价抑制剂中(表1)例如,Fumagillin通过与活性位点His231的共价结合不可逆地抑制甲硫氨酸肽酶2 (MetAP2)。Carfilzomib是四肽环氧酮天然产物epoxomicin的类似物。它与20S蛋白酶体的催化活性苏氨酸残基不可逆地共价结合,被批准用于临床治疗多发性骨髓瘤。环氧化物的氧原子质子化对于靶头亲电性的激活非常重要。双环[1.1.0]丁烷(BCB)是一种高度应变的碳环,具有独特的蝴蝶形结构。(表1)虽然BCB环系统通常是热稳定的,但在桥头碳上引入吸电子基团(EWG)将促进应变驱动的亲核加成。Baran团队证实了BCB砜可选择性地与半胱氨酸硫醇结合。
6.3硼酸以及硼酸酯
硼酸和硼酸酯的硼中心与丝氨酸和苏氨酸残基的羟基形成可逆的共价键。(表2)硼替佐米和伊沙佐米已被临床批准用于治疗多发性骨髓瘤。这些药物通过靶头的硼中心与催化活性的苏氨酸残基结合来抑制20S蛋白酶体。
6.4乙二醛类
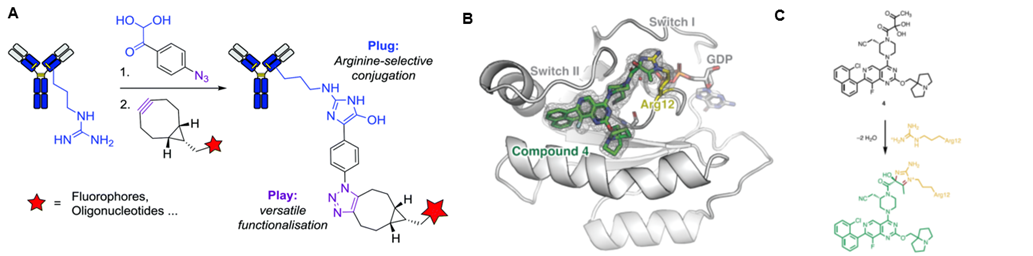
Dovegan团队以4-叠氮苯基乙二醛(APG)作为亲电性弹头,成功实现了对天然抗体中的精氨酸残基的选择性共价修饰(图11A)。APG和精氨酸的胍基之间的选择性反应让该团队轻松在单克隆抗体曲妥珠单抗上引入了叠氮基。Zhang小组使用α,β-二酮酰胺作为靶向精氨酸的靶头,首次实现了对K-Ras(G12R)突变体的选择性修饰(图11B)。因此通过合理设计乙二醛的类似物,或许能实现目标蛋白赖氨酸残基的选择性修饰。
6.5 其他用于TCI设计和化学选择性生物聚合的化学策略
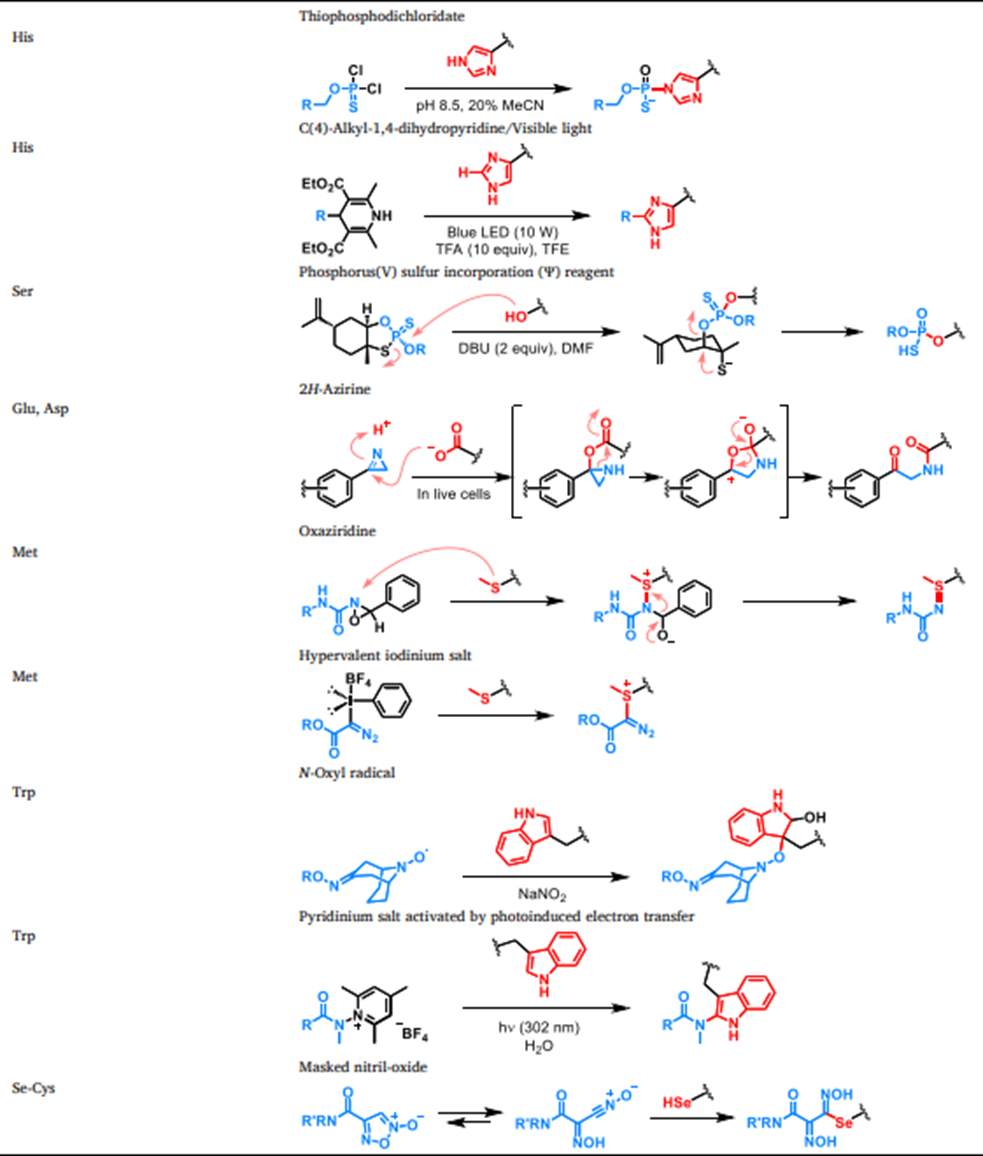
目前共价药物的研究通常利用蛋白质氨基酸残基的亲核反应性设计靶头,例如半胱氨酸、赖氨酸和活化的丝氨酸/苏氨酸。然而,对其他氨基酸侧链的化学选择性共价修饰是扩大蛋白质共价配体库的重要方向。图12总结了蛋白质氨基酸残基化学选择性生物偶联的最新进展。
结论
本篇综述简单介绍了用于选择性修饰目标蛋白的共价靶头,归纳了其结构特征及其类别。从药物研发的角度,TCI靶头应在体内生物条件下保持化学和代谢的稳定,同时保持对目标蛋白质的足够反应性。虽然目前大多数批准的TCIs使用Michael受体型靶头,但随着新的亲电靶头结构被不断报导,我们相信后者在未来将展现出独特的临床价值。
可逆共价抑制剂作为另一种宝贵的治疗策略,在实现精确靶向的同时,也减轻了脱靶反应引起的副作用。本文中提到的所有靶头皆为亲电性靶头,其自身结构和邻位取代基的电性,所靶向氨基酸残基的亲核性都会影响靶头的反应活性和化学选择性。而想要提高TCI对目标蛋白的靶向性,则需要增加靶蛋白结合口袋位置与TCI分子之间的非共价相互作用。
除此以外,积极地对靶头的取代基进行构效研究也有助于增强靶头的共价反应活性,甚至得到可逆的共价抑制剂。除了亲核性较强的氨基酸,其他与癌症、疾病和生理功能密切相关的氨基酸诸如精氨酸和甲硫氨酸也是共价靶头的研究重点,对后者的探索将扩大共价靶头的化合物库,丰富TCI的靶点种类并促进以TCI为基础的化学疗法例如化学蛋白降解剂、PPI抑制剂和抗体-药物偶联物的开发。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























