突破性治疗药物是指药物临床试验期间,用于防治严重危及生命或者严重影响生存质量的疾病,且尚无有效防治手段或者与现有治疗手段相比有足够证据表明具有明显临床优势的创新药或者改良型新药等。
为了适应不断增长的药物需求并跟上中国制药领域的快速发展,为鼓励创新和满足临床急需,2020年7月,国家药监局发布《突破性治疗药物审评工作程序(试行)》文件的公告,申请人可在Ⅰ、Ⅱ期临床试验阶段,通常不晚于Ⅲ期临床试验开展前申请突破性治疗药物。
从CDE的公示信息可以看出2020年至今已有上百款药物纳入了突破性治疗品种。药品涵盖了当下较为热门的创新疗法,如CAR-T疗法、ADC、双特异性抗体等。本文就11月刚刚批准纳入突破性治疗药物的品种进行整理,以此了解CDE对突破性治疗药物的要求,为企业申请突破性治疗品种提供参考。
01
pCAR-19B
批准时间:2023年11月1日
药品名称:pCAR-19B细胞自体回输制剂
申报企业:重庆精准生物技术有限公司
靶点:CD19
适应症:CD19阳性急性淋巴细胞白血病
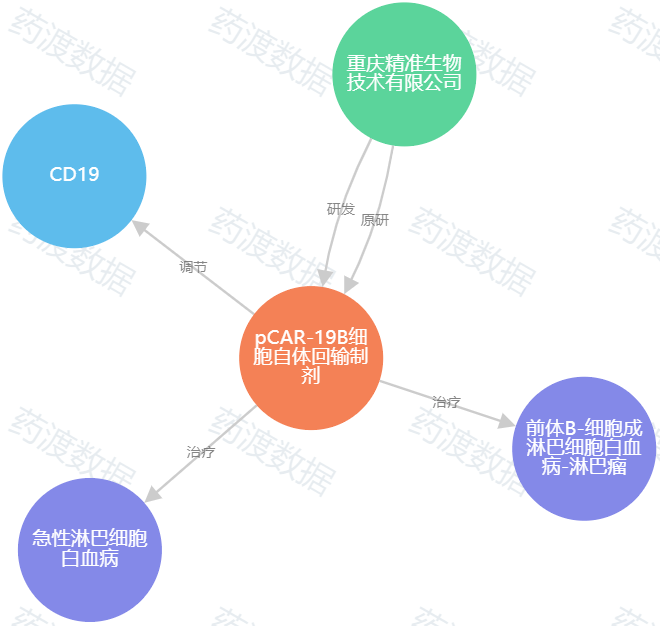
pCAR-19B是一款由重庆精准生物自主研发的人源化CD19 CAR-T,能够显著降低CAR-T细胞免疫原性,延长CAR-T细胞在体内的存续时间。据了解,pCAR-19B是由重庆精准生物采用其Precision CAR设计策略打造出的更为有效的CAR-T产品。pCAR-19B细胞自体回输制剂已经于2019年获儿童、青少年急性淋巴细胞白血病(B-ALL)适应症注册临床默示许可。它是国内首 个针对该适应症进入到II期注册临床的CAR-T产品,也是国内首 个针对该适应症的人源化设计的CAR-T产品。
该产品于2019年11月启动I期注册临床试验,纳入9例符合标准的难治性或多线治疗后复发的儿童或青少年B-ALL患者,且超过一半受试者为高瘤荷患者。数据显示,9例患者均获得完全缓解(CR),总体缓解率达100%,且首次达到完全缓解(CR)的患者微小残留病变(MRD)也均为阴性;无剂量限制性毒性(DLT)和治疗相关死亡事件发生,总体安全性和耐受性良好。2022年,pCAR-19B产品开始关键Ⅱ期注册临床试验,该临床试验为多中心临床试验,目标入组人数为100人,主要目标为评价pCAR-19B细胞自体回输制剂治疗CD19阳性复发/难治性B细胞急性淋巴细胞白血病的3个月客观缓解率(ORR)。
02
谷美替尼片
批准时间:2023年11月10日
药品名称:谷美替尼片
申报企业:上海海和药物研究开发股份有限公司
靶点:c-Met
适应症:既往接受免疫治疗(抗PD-1/PD-L1抗体)和含铂双药化疗后(联合用药或序贯用药)出现疾病进展的驱动基因阴性且伴有MET过表达的局部晚期或转移性非小细胞肺癌(NSCLC)
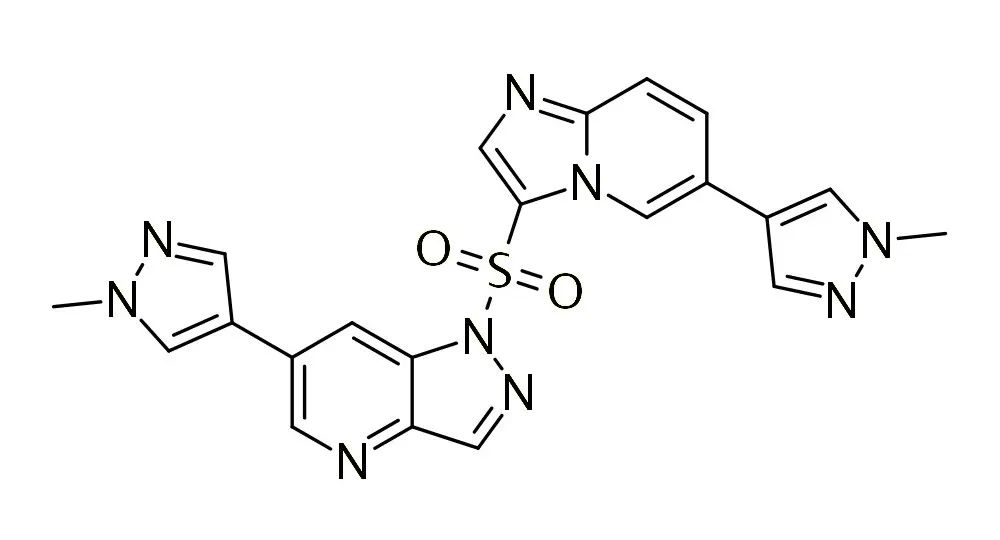
谷美替尼(SCC244)是一种高选择性c-Met抑制剂,该药能够选择性抑制c-Met激酶活性,进而抑制肿瘤细胞的增殖、迁移和侵袭,在携带MET外显子14跳跃突变的非小细胞肺癌患者中,表现出持久疗效。临床研究结果显示,谷美替尼片具有优良的药代动力学特性以及良好的安全性和耐受性,在人体中药物半衰期长,稳态谷浓度高,有利于靶点的持续抑制。
据CDE网站显示,该药在2021年就曾纳入了突破性治疗品种。2023年3月,国家药品监督管理局正式批准海益坦?(谷美替尼片)适用于治疗具有间质-上皮转化因子(MET)外显子14跳变的局部晚期或转移性非小细胞肺癌患者。
一项开放、国际多中心的单臂II期研究GLORY试验(NCT04270591),主要评估了谷美替尼治疗MET外显子14跳变的局部晚期或转移性NSCLC患者的有效性和安全性。在该试验中,总共入组了84例患者,其中79例具有中心实验室确认的MET外显子14跳变的非小细胞肺癌患者,初治患者44例,经治患者35例。在所有人群中,由盲态独立影像评估委员会(BIRC)评估的总体客观缓解率(ORR)为66%。谷美替尼起效迅速,绝大部分在首次肿瘤评估中达到缓解,中位起效时间为1.4个月。中位缓解持续时间(DOR)为8.3个月;中位无进展生存期(PFS)为8.5个月;中位总生存期(OS)为17.3个月;疾病控制率(DCR)为84%。在初治患者中ORR为71%,中位DOR为15.0个月;中位PFS为11.7个月;中位OS尚未达到。在经治患者中,ORR为60%,中位DOR为8.3个月;中位PFS为7.6个月。14例脑转移患者中,有13例被纳入疗效分析集。
结果显示,这些脑转移患者的总体ORR高达85%,观察到了令人鼓舞的颅内抗肿瘤作用。在安全性方面,整体安全可控。最常报告的治疗相关不良事件包括水肿、低白蛋白血症、头痛、食欲不振和恶心,这些是接受MET抑制剂治疗的患者中已知的常见不良反应。
03
BIVV020注射液
批准时间:2023年11月14日
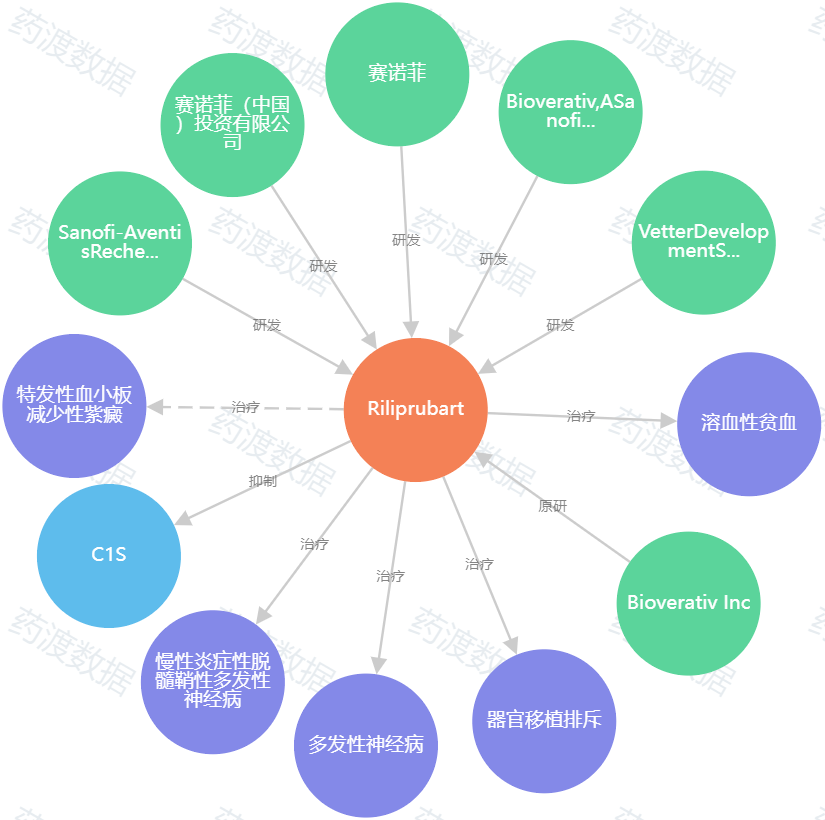
药品名称:BIVV020注射液
申报企业:赛诺菲(中国)投资有限公司
靶点:补体C1s
适应症:慢性炎性脱髓鞘性多发性神经根神经病(CIDP)
CIDP是一种罕见的自身免疫疾病,会影响到大脑和脊髓外的外周神经。罹患这种疾病的患者,其保护神经的髓鞘会出现损伤,导致肌肉出现麻木、虚弱、疲劳等症状。随着病情的持续,CIDP的症状会进一步加重,极大地限制了患者的运动能力,并降低他们的生活质量。
BIVV020(SAR445088)是一种人源化IgG4单克隆抗体,可与特定的丝氨酸蛋白酶 C1选择性结合,从而阻止下游酶级联的激活,导致C3转化酶的激活和膜攻击复合物的形成。补体途径也激活促进巨噬细胞募集、炎症和细胞裂解,同时使凝集素和旁路途径在功能上保持完整,以便宿主防御。在补体介导疾病的治疗中,具有潜在的临床益处。
其在93名健康参与者中进行了单次和多次给药SAR445088的I期、首次人体双盲、随机、安慰剂对照、剂量递增试验,结果表明:SAR445088耐受性良好,在人类单次给药和多次给药后,均具有良好的PK和PD谱(药代动力学和药效学)。
这些发现支持对经典补体介导性疾病患者进行进一步的临床研究。根据中国药物临床试验登记与信息公示平台官网,赛诺菲正在开展SAR445088治疗慢性炎性脱髓鞘性多发性神经根神经病(CIDP)成人患者的II期概念验证研究。该研究的主要目的为评价SAR445088在标准治疗(SOC)经治、SOC难治和SOC未治人群三个CIDP患者亚群中的有效性,以及评价SAR445088在CIDP长期治疗中的安全性和耐受性。
04
Vamorolone口服混悬液
批准时间:2023年11月14日
药品名称:Vamorolone口服混悬液
申报企业:Santhera Pharmaceuticals (Switzerland) Ltd;曙方(上海)医药科技有限公司;Purna Pharmaceuticals SA
靶点:GR(糖皮质激素受体)
适应症:杜氏肌营养不良症
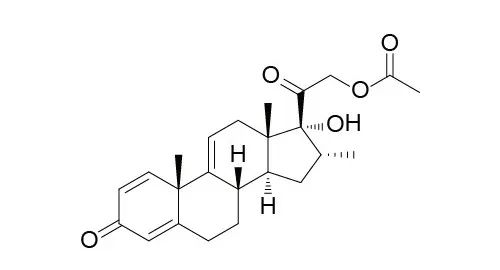
在杜氏肌营养不良症(DMD)患者中,类固醇药物的抗炎症效果是介导药物疗效的主要机制,但是类固醇的其它活性会带来影响患者生活质量的副作用。Vamorolone与普通类固醇药物的区别在于——能够有选择性地激活类固醇的某些信号通路,能够在保持类固醇对DMD疗效的同时减少副作用的产生。
Vamorolone获得美国与欧洲用以治疗DMD的孤儿药资格,并获得美国FDA快速通道资格与罕见儿科疾病认定。2023年10月26日,美国FDA批准了Vamorolone口服混悬液40 mg/mL的上市申请,用于治疗2岁及以上杜氏肌营养不良(DMD)患者。Vamorolone最初由ReveraGen BioPharma开发。2018年,Santhera公司获得了该药的全球研发许可(除日本和韩国以外)。2022年1月,曙方医药与Santhera公司宣布就vamorolone达成独家授权协议,从而获得该产品在大中华区用于DMD及其他罕见病适应症的独家开发和商业化权益。
FDA批准是基于一项关键2b期临床试验VISION-DM积极的临床数据。试验评估了vamorolone治疗DMD患者的疗效和安全性,结果达到了主要终点,试验组在治疗24周后由卧位至站立所需时间(TTSTAND,DMD儿童患者疾病恶化的第一项功能性指标)优于安慰剂组,且差异有统计学意义。另外,该研究许多次要终点的结果也与主要终点结果一致。Vamorolone在试验中表现出良好的安全性和耐受性,最常见的不良事件是头疼、呕吐和维生素D缺乏,但不良事件的严重程度一般为轻至中度。
05
BRII-179
批准时间:2023年11月22日
药品名称:BRII-179 (VBI-2601) 注射液
申报企业:腾盛博药医药技术(北京)有限公司
适应症:乙肝治疗性疫苗
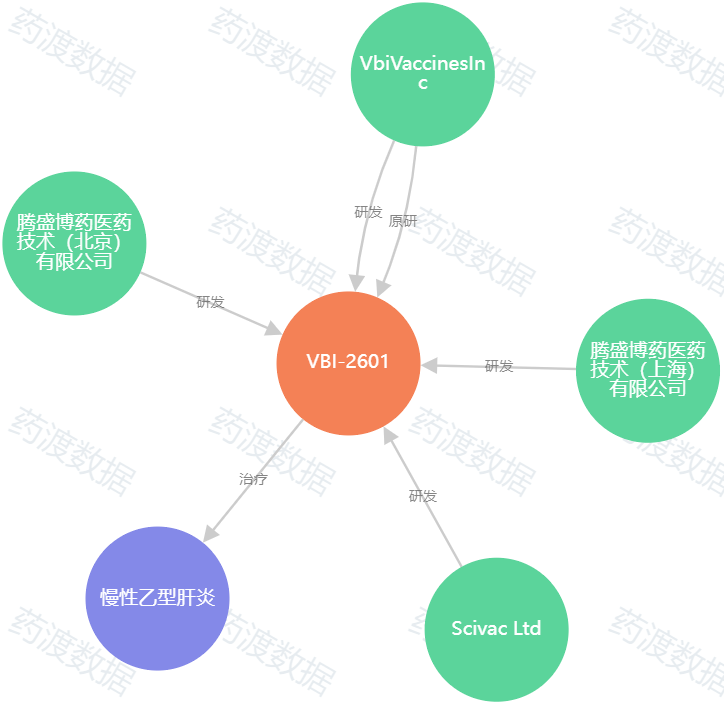
BRII-179是一种包含3种HBV表面抗原组成的治疗性疫苗,作用机制为靶向增强B细胞和T细胞特异性免疫,从而实现慢性HBV感染功能性治愈的目标。2018年腾盛博药从VBI Vaccines引进BRII-179,获得了在大中华区的商业化权益。自2023年7月起,该公司还将BRII-179的独家许可扩展至全球。
在现有PEG-IFNα(聚乙二醇干扰素α)基础上联合BRII-179治疗,总体安全且耐受性良好,其不良事件与既往报道的PEG-IFNα治疗和BRII-179的不良事件相似。治疗结束12周,与安慰剂+PEG-IFNα组相比,BRII-179+PEG-IFNα组的HBsAg(乙肝表面抗原)清除率更高。在治疗结束24周仍观察到该HBsAg清除率方面的差异,这种差异一直维持到第36周。临床研究还发现,在第24周,联合给药组的HBsAg血清学转换率显著高于安慰剂+PEG-IFNα组。
06
恩沃利单抗
批准时间:2023年11月24日
药品名称:恩沃利单抗注射液(恩维达?)
申报企业:四川思路康瑞药业有限公司;江苏康宁杰瑞生物制药有限公司
靶点:PD-L1
适应症:恩维达?联合仑伐替尼治疗既往至少一线含铂化疗失败或不能耐受的非微卫星高度不稳定(MSI-H)/ 非错配修复基因缺陷型(dMMR)晚期子宫内膜癌。
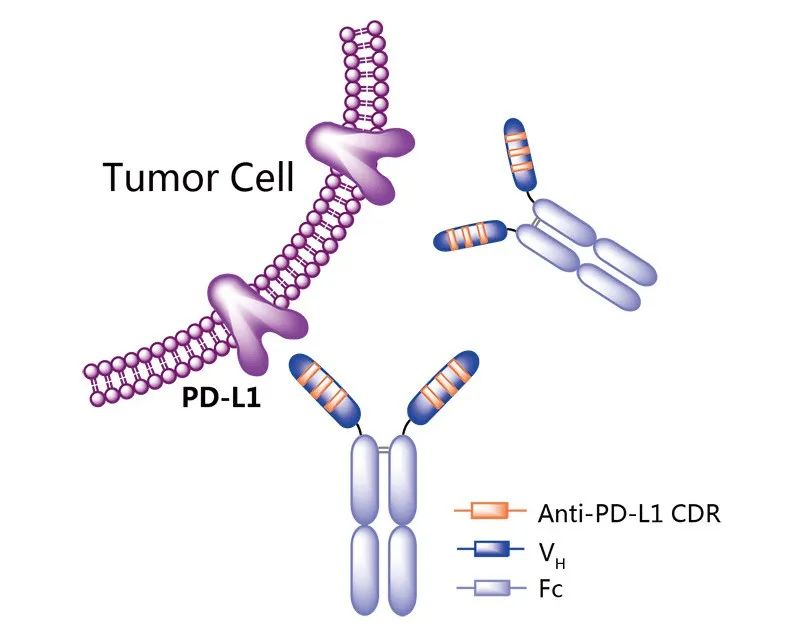
恩沃利单抗是一种由单域抗体(sdAb)和Fc段组成的单特异性抗体,分子量是完整抗体分子量的一半,这使其具有增强的穿透性,同时具有完整的抗原结合能力。此外,Fc-介导的效应功能在恩沃利单抗中被削弱,以限制其接触免疫系统并避免意外的不必要免疫反应。2021年,NMPA批准上市适应症为不可切除或转移性微卫星高度不稳定(MSI-H)或错配修复基因缺陷型(dMMR)的成人晚期实体瘤患者的治疗。
仑伐替尼是一种口服多靶点激酶抑制剂,可选择性抑制血管内皮 生长因子(VEGF)受体的激酶活性,以及抑制其他促血管生成和致癌通路相关的受体酪氨酸激酶(RTK)活性,包括成纤维细胞生长因子(FGF)受体,血小板衍生 生长因子(PDGF)受体,转染重排(RET)等。免疫疗法与抗血管生成靶向药物联用能显著提高患者获益。
早前,仑伐替尼原研产品已在美国获批与PD-1抑制剂联合用于治疗特定晚期子宫内膜癌患者。而恩沃利单抗与仑伐替尼联用方案的优势在于恩沃利单抗为皮下注射给药,仑伐替尼为口服片剂,二者均用药便捷,有利于提高患者依从性,实现肿瘤慢病化管理。2023年10月,恩维达?联合仑伐替尼治疗复发性子宫内膜癌患者的Ⅲ期临床研究(KN035-US-004)获得FDA的新药临床试验批准。
07
JMKX001899
批准时间:2023年11月29日
药品名称:JMKX001899片
申报企业:浙江杭煜制药有限公司
靶点:KRAS G12C
适应症:既往至少接受过一种系统性治疗的KRAS G12C突变的晚期或转移性非小细胞肺癌患者
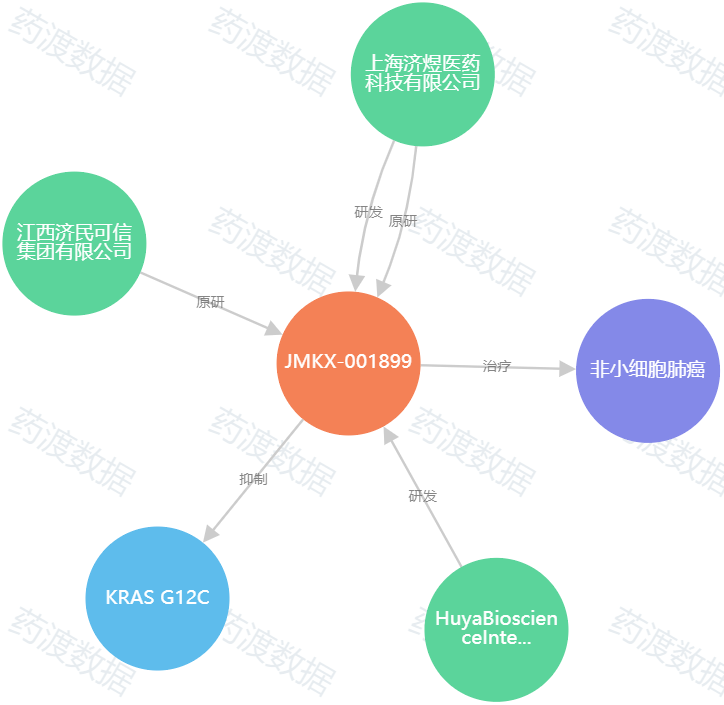
JMKX001899是济民可信集团研究院研发的一款口服新型小分子化合物,与KRAS G12C突变生成的半胱氨酸共价结合后,更倾向与鸟苷二磷酸(GDP)的结合,导致鸟苷三磷酸(GTP)与KRAS的亲和力降低,通过将KRAS G12C突变体特异性不可逆地锁定在非激活的GDP结合状态,从而抑制肿瘤细胞增殖活性,达到抗肿瘤效果。
一项开放标签、多中心 I/II 期临床研究仍在进行之中,以评估JMKX001899片剂在携带KRAS G12C突变的晚期或转移性实体瘤患者(Ia 期)和非小细胞肺癌患者中的安全性、耐受性、药代动力学和抗肿瘤活性。
小结
CDE对纳入突破性治疗药物审评程序的品种会采取一系列支持政策,有助于药品走上“加速审批”的快车道,对于制药公司是缩短研发时间、简化试验设计、提升公司知名度和融资的利器。但想要拿到“入场券”还需要药物具有突破性的临床价值。需要注意的是突破性治疗仅针对创新药或者改良型新药,只针对严重危及生命和严重影响生存质量的两类疾病,且临床的安全有效性数据得到认可。
在目前内卷严重的制药行业,申请突破性治疗药物或许将成为药企的一条新出路。期待有更多的突破性治疗药品能加速上市,进一步满足中国重大疾病的临床治疗需求,为无药可医的罕见病、肿瘤等患者带来福音 。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























