肿瘤抑制蛋白p53是一种转录因子,在调节细胞周期进展和细胞死亡中起着关键作用——P53诱导其自身抑制剂(人类双微体-2、HDM2)的转录。
HDM2与p53结合,抑制其转录活性,将p53蛋白引进细胞质,并通过E3泛素途径将其靶向蛋白酶体降解。
HDM2通过三种机制抑制p53活性:
1)充当E3泛素连接酶以促进p53降解;
2)结合并阻断p53转录活化结构域;
3)将p53从细胞核输出到细胞质。
在某些类型的癌症(骨肉瘤、软组织、肺癌和脑瘤等)中已经观察到,HDM2的过表达使P53失活。HDM2-p53蛋白-蛋白相互作用的中断(PPI)能解除p53的负调控,使其发挥抗增殖和促凋亡功能。因此,使用HDM2?p53 PPI抑制剂已经成为治疗人癌症(WT p53)的一种有吸引力的方法。
每一篇JMC中的Drug Annotation都是一个新药开发的小故事,本篇文章想分享一下默克公司的MK4688的改造故事。
两种蛋白接触的表面具有large, flat的疏水界面,使得蛋白蛋白抑制剂与经典的口服药物相比具有分子量更大、更亲脂的特点,这类分子特性导致临床需要高剂量和PK具有可变异性,从而降低了分子最终成药的可能性。
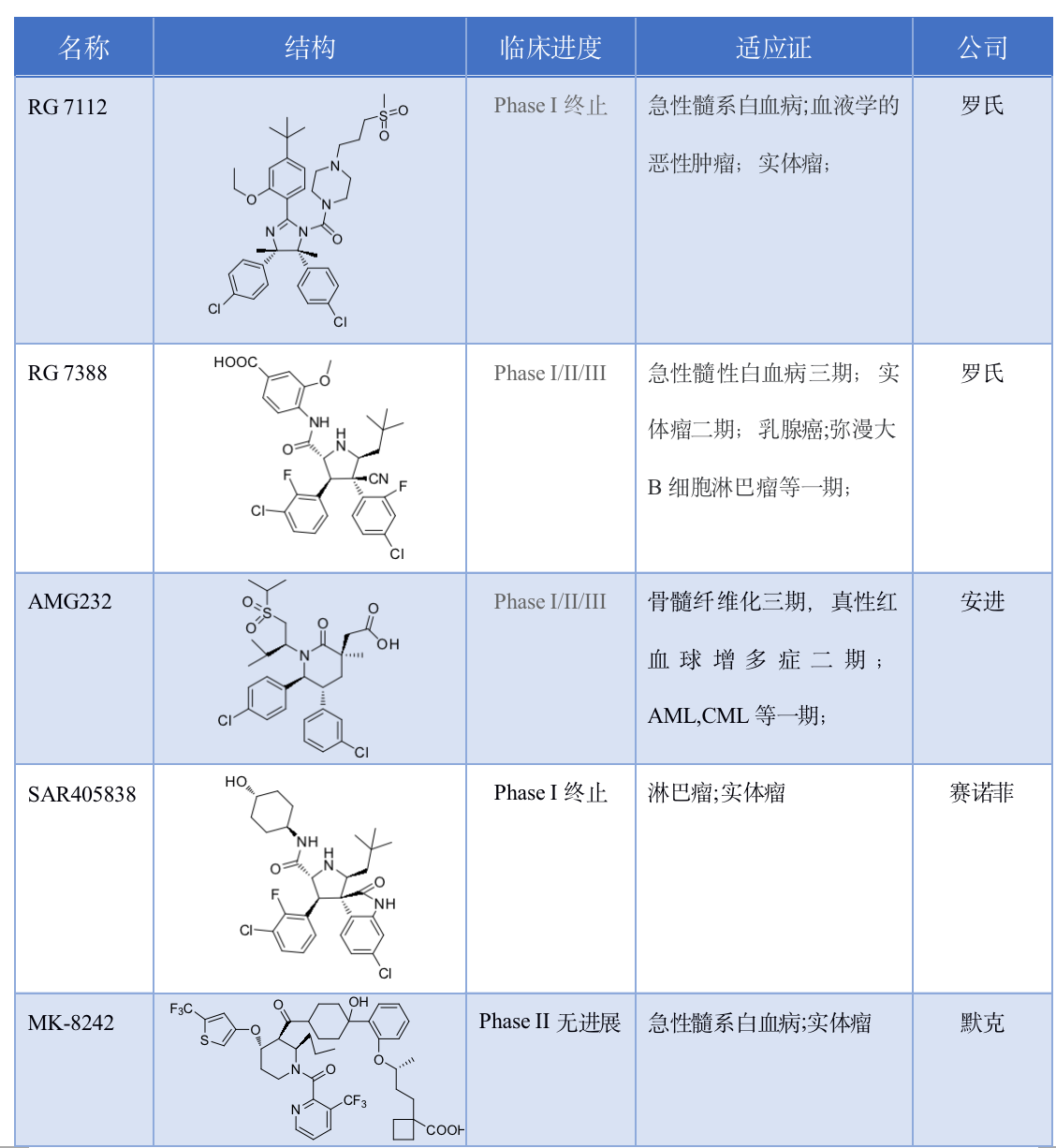
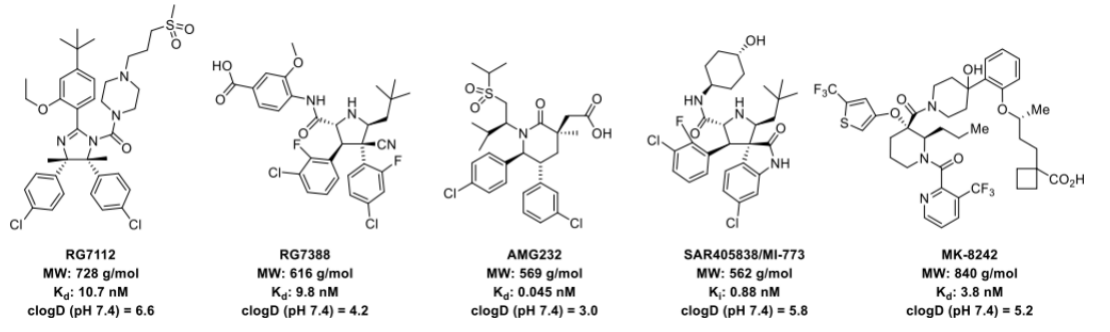
图1 几类不同骨架的HDM2抑制剂及其理化性质、活性和临床进展
HDM2与p53的相互作用主要是通过疏水界面介导的,其中位于p53的N端α螺旋上的Phe19、Trp23和Leu26残基与HDM2蛋白表面的疏水口袋产生相互作用。
尽管PPI界面具有这一特点,仍有一些HDM2抑制剂进入了临床开发中,图1这些抑制剂都具有一些特点:高亲脂性(Clogp较大)、高分子量(MW)、需要高剂量才能达到临床需要的有效性。
在一期临床中的AML(急性髓系白血病)试验,RG7112显示了可变异性的暴露,在MTD剂量(1500 mg,BID)下,只有37%的患者达到了实现有效性所需要的最小暴露量。因此,提高HDM2抑制剂的类药性非常重要。
第二代的RG7388, SAR405838, AMG232完善了分子的理化性质,目前处于临床研究阶段。MK-8242分子具有高MW,高clogp,较高的推荐剂量(2期剂量为每天0.8 g);默克公司最初通过高通量筛选新的骨架分子以期望得到分子量低、剂量也低的HDM2-P53抑制剂。
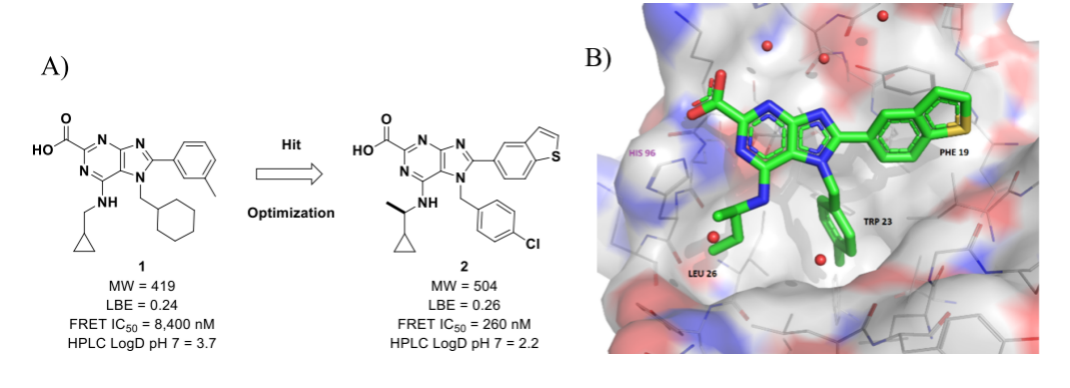
图2 hit的优化过程及2和HDM2蛋白的共晶 (PDB 7NA1)
通过高通量筛选得到化合物1,这是一个分子量低、带嘌呤骨架的小分子化合物,对hit的优化集中在嘌呤母核的C6、N7和C8上,环己亚甲基替换为对氯苄基、间苯基改为苯并噻吩、NH的α位碳引入手性甲基;最终活性提高了30倍,并提高了LBE(配体结合效率);通过观察化合物2和HDM2蛋白的X-ray晶体结构,2的嘌呤骨架具有α螺旋的特征,高效的将取代基伸入3个已报道过的HDM2蛋白表面的hot spot结合口袋中,C6烷基胺占据Leu26口袋,N7的苄基填充Trp23口袋,C8苯并噻吩占据了Phe19口袋。C2羧酸与HDM2的His96侧链发生关键的静电相互作用,类似于AMG232的羧酸取代基的作用。
默克公司的优化策略是在尽量不增加分子量的基础上提高活性、提高成药性。
图3 取代基团优化过程
根据图2的共晶,羧酸位于His96口袋、苯并噻唑在Phe19口袋、氯苯在Trp23口袋、环丙甲基在Leu26口袋;从图3看出,先导2中的环丙基改为环丁基、氯苄基改为三氟甲基苄基提高了与leu26和Trp23的疏水接触,观察Phe19口袋的空腔可以减小取代基团的大小,得到的化合物23,活性提高了17倍;最终把红色片段优化到苯环手性取代的吗啉环得到化合物27,活性达到1.8 nM;根据NMR-based模型,蓝色区域的Trp23口袋更适合饱和脂肪环占据,且引入了手性发现化合物33,活性达到2.2 nM,把27和33的最优取代片段整合到一块得到了化合物15,活性达到0.2 nM.
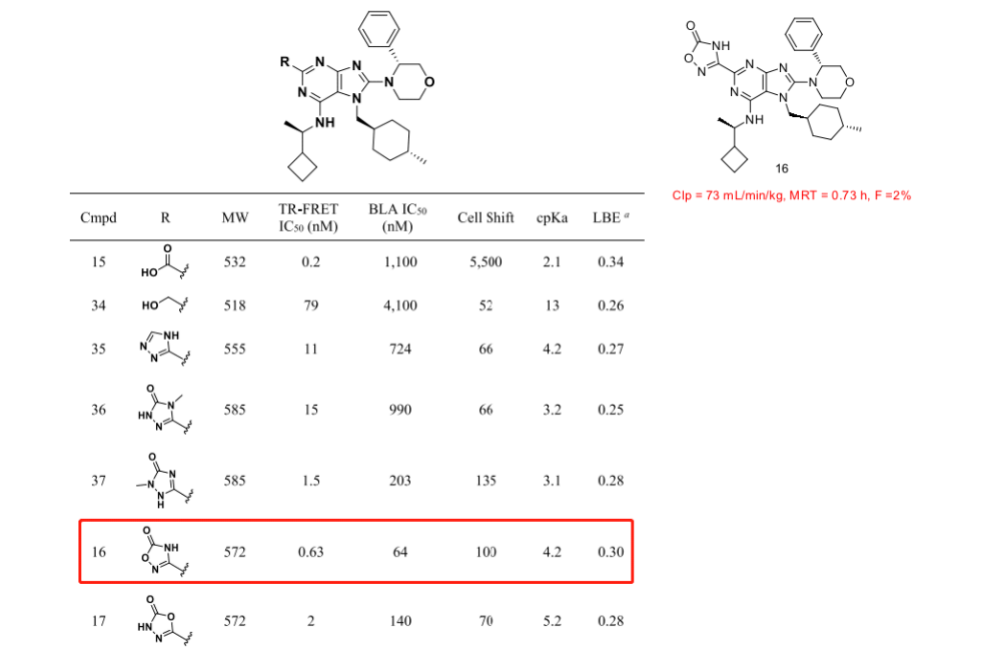
图4 C2位羧酸的生物电子等排改造
在取得了优异的TR-FRET的 IC50活性后,化合物15的细胞活性BLA IC50只有1.1μmoL,降低了5500倍,这种情况很可能是因为嘌呤羧酸部分(计算pKa为2.1)的高酸度导致被动渗透性差,由于C2羧酸与HDM2的His96侧链发生了关键的氢键作用,默克公司做了羧酸的生物电子等排来改造结构。最终得到图4中的化合物16,细胞活性进入百纳以内。但是化合物16在大鼠中的清除率高达73 mL/min/kg,生物利用度也只有2%,仍需接着改。默克研发人员认为,高的亲脂性导致了脱靶和较差的PK,他们后续采取了一种策略,专注于降低总体log D和提高分子的LLE。
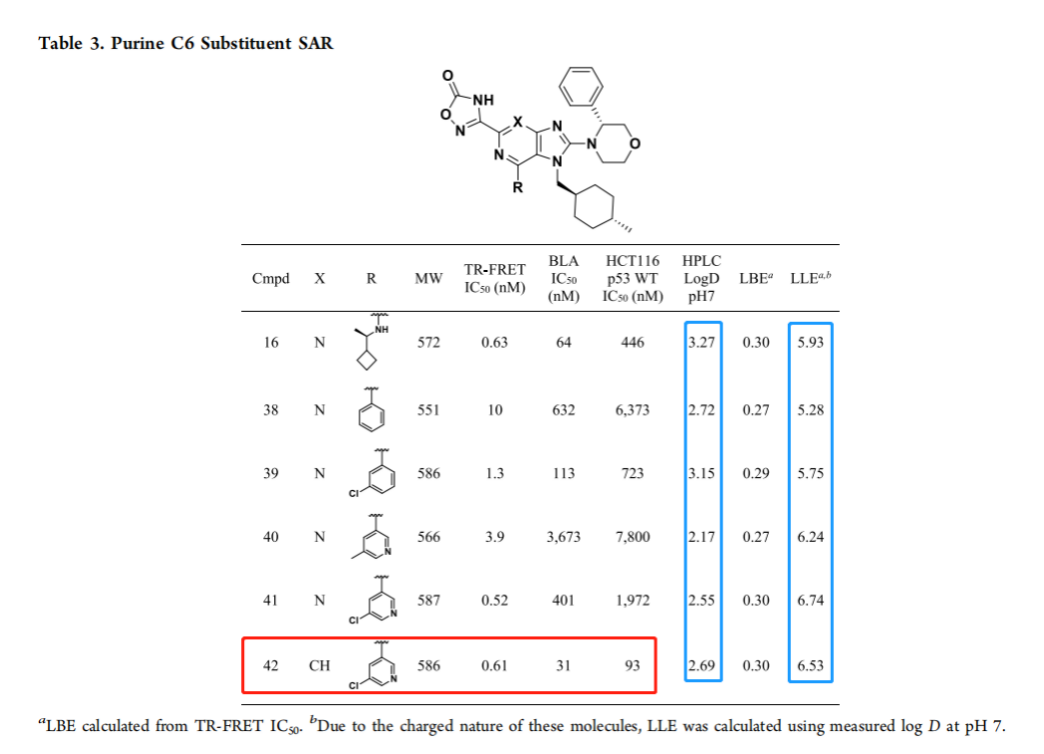
图5 C6位的构效关系
从图5可以看出,嘌呤骨架中的X改为C,可以降低分子的酸性,提高PKa,此外,基于嘌呤衍生化合物的NMR-based模型,嘌呤N3和N9都位于HDM2表面的疏水区域,改成碳原子也有利于疏水结合;最终间氯吡啶环的引入得到了化合物42,与16相比,LLE由5.93提高至6.53,LogD由3.27降低至2.69. 但是化合物42的PK仍然有缺陷。
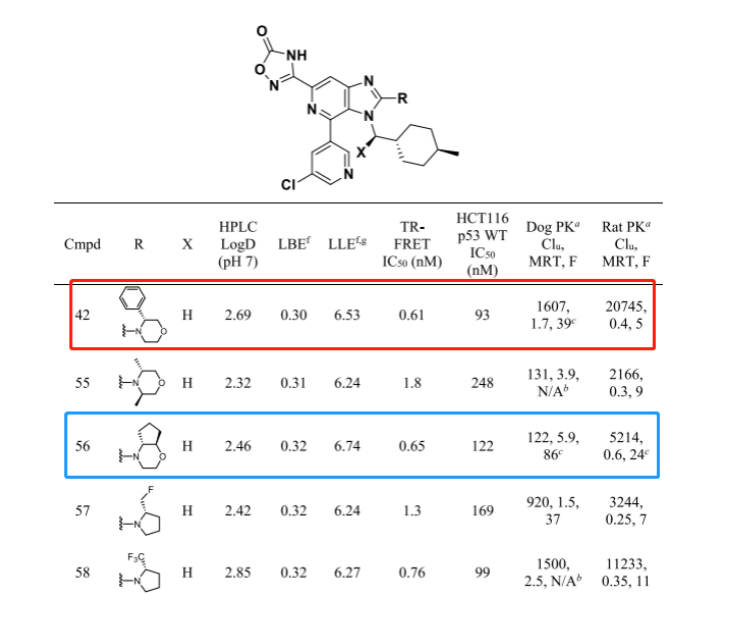
图6 咪唑嘧啶骨架上对PK优化
对PK继续优化,42的MRT在两种种属中都很短(在大鼠和狗中分别为0.4和1.7 h),这表明需要优化来实现低剂量给药,MetID对42的研究指出亲脂取代的吗 啡啉和环己基甲基是易代谢位点。因而把甲基去掉,同时封闭吗啉环上的易代谢位点,得到了化合物56,在犬和大鼠中的PK均比较优秀。
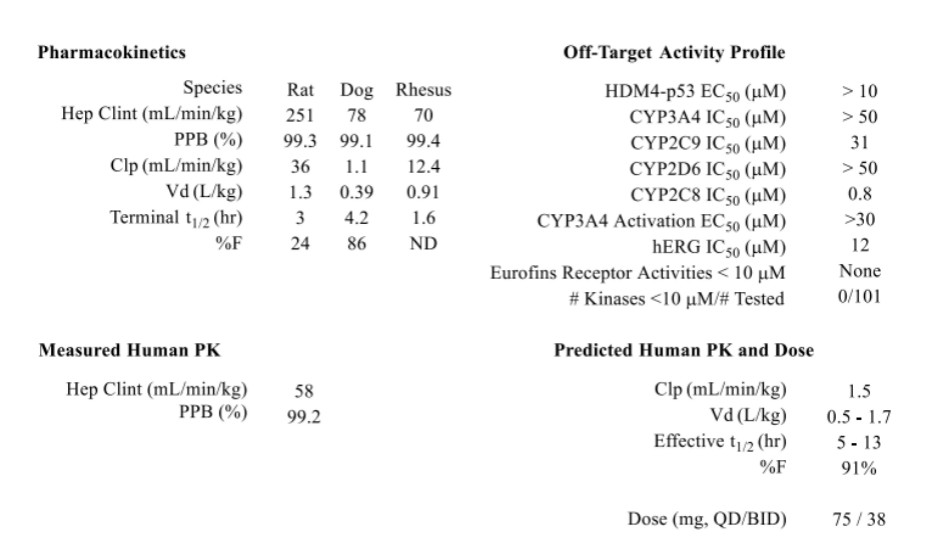
图7 大鼠、狗和恒河猴的临床前PKs
最后,详细的在大鼠、犬和恒河猴体内进行了单次静脉或口服给药后的PK研究。化合物56在体外和体内清除存在物种差异,其中犬清除低,猴和大鼠清除中等;此外56在犬中有很好的口服生物利用度,在大鼠中具有中等的生物利用度,这可能是由于肝脏首过效应的差异。尽管存在这些差异,但在不同物种间观察到很强的体外/体内相关性,支持了一个可靠的人体内PK预测。根据默克的第一代HDM2抑制剂MK-8242的临床经验,设定了25 μM h的人的目标暴露量(AUC)。将这些分析与预测的人药代动力学相结合,预测56给药下人的剂量为75 mg QD (38 mg BID)。与MK-8242达到相同暴露水平所需的剂量相比,以及与其他第一代HDM2抑制剂疗效所需的剂量相比,这一低剂量代表了显著的改善。化合物56也被称为MK-4688,目前仍在临床前继续进行安全性研究。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























