在医药领域这个“苟日新,日日新,又日新”的环境中,创新和突破是永恒的主题。
2024年1月5日,FDA批准Zelsuvmi?(Berdazimer外用凝胶,10.3%),用于治疗成人和一岁及以上小儿的传染性软疣,成为首 个全球治疗软疣感染的新型药物,也是治疗这种具高度传染性的病毒性皮肤感染的首 个局部使用处方药。
那么2024年FDA还有望批准哪些药物呢?
注:1992年开始的《处方药使用者付费法案》(The Prescription Drug User Fee Act ~ PDUFA),被业界称之为“美国现代新药审评的基石”,开创了新药审评审批的新纪元,且直至今日,仍在更新使用当中。
01
Zolbetuximab
原PDUFA日期:2024.01.12
Zolbetuximab是由德国生物技术公司Ganymed公司开发的一款靶向CLDN18.2蛋白的具有人类IgG1恒定区的小鼠嵌合单抗药物。
2016年10月,安斯泰来以14亿美元收购Ganymed,将Zolbetuximab收入囊中。
CLDN18.2在很多癌症中过表达,尤其是胃癌,HER2在胃癌患者中的阳性率不到20%,而CLDN18.2在相关癌症中的阳性率超过50%,成为胃癌领域热门靶点。
目前尚无CLDN18.2靶向药获批上市,此前,Zolbetuximab一直是CLDN18.2靶向药的领头羊,2023年7月份,安斯泰来向FDA递交了Zolbetuximab的新药上市申请(NDA),并且获FDA优先审评资格,处方药使用者付费法案(PDUFA)日期为2024年1月12日。
然而,2024年1月9日,安斯泰来宣布了FDA于1月4日发布的一份完整的回复函:FDA表示无法在2024年1月12日PDUFA行动日期之前批准BLA,原因是第三方生产工厂存在未解决缺陷[1]。
这一黑天鹅事件耽误了zolbetuximab成为首 款上市的靶向CLDN18.2 的胃癌药,使其独占市场的时间越来越少。
毕竟除了zolbetuximab,临床上还有很多在研的CLDN18.2靶向药,如创胜集团的osemitamab和奥赛康的ASKB589等。
不过,好消息的是FDA尚未对zolbetuximab的临床数据(包括疗效或安全性)提出任何担忧,也没有要求进行额外的临床研究。
而且安斯泰来正在与FDA和第三方制造商密切合作,以制定时间表,以快速解决该机构的反馈。相信zolbetuximab今年还是有很大希望被FDA获批上市。
Zolbetuximab在国内的上市申请已于2023年8月1日获得受理,用于治疗胃癌及胃食管交界处(GEJ)腺癌,成为中国首 个上市申请的CLDN18.2单抗。
02
Lifileucel
PDUFA日期:2024.02.24
Lifileucel是由Iovance Biotherapeutics研发的一种TIL(Tumor Infiltrating Lymphocyte Cell,肿瘤浸润淋巴细胞)疗法,正在临床上开展多项单药或者组合疗法,用于治疗在既往抗 PD-1/L1 治疗和靶向治疗(如适用)期间或之后进展的晚期黑色素瘤患者(图1)。

图1. Lifileucel的开发进展
TIL 疗法属于免疫细胞疗法,是由CD8+T细胞、CD4+T细胞、B细胞、NK细胞与γδT细胞等组成的异质性细胞群体,主要通过细胞毒性CD8+T细胞对肿瘤细胞进行直接杀伤。
TIL细胞疗法技术流程是将肿瘤组织中的T细胞分离出,在体外进行刺激扩增后,回输到患者体内,从而扩大免疫应答,治疗原发或继发肿瘤。
2023年3月24日,Iovance宣布Lifileucel已成功完成其滚动上市申请提交工作,PDUFA日期为2023年9月11日。
然而2023年9月份,FDA 表示他们没有足够的资源在计划于2023年9月11日举行的周期后期审查会议之前审查最近对Lifileucel正在进行的BLA审查的信息请求的回应。
FDA将PDUFA日期延长至2024年2月24日,但同意与Iovance合作,加快剩余的审查,以获得可能更早的批准日期,如果获得批准,Lifileucel将成为第一个也是唯一一个针对晚期黑色素瘤患者的TIL疗法,也是第一个针对实体瘤癌症的一次性细胞疗法[2]。
此前,FDA 已于2023年5月在将lifileucel纳入优先审评,而且也授予 lifileucel 用于晚期黑色素瘤的再生医学先进疗法(RMAT)称号。此次PDUFA 日期的延长不会影响优先审查状态或 RMAT 指定。
在2023年的ESMO大会上,Lifileucel带来了治疗晚期粘膜黑色素瘤患者积极的临床结果:独立审查委员会(IRC)使用RECIST v1.1评估的ORR为50%(95% CI:21%–79%)。在中位研究随访35.7个月时,未达到(NR)中位缓解持续时间(DOR),中位无进展生存期(PFS)为NR(95%CI:1.4个月–NR),中位总生存期(OS)为19.4个月(95%CI:7.9个月–NR)。治疗中出现的不良事件(TEAE)与淋巴细胞清除化疗和白细胞介素-2 (IL-2) 的已知安全性一致[3]。
03
Roluperidone
PDUFA日期:2024.02.26
Roluperidone是由Minerva Neurosciences从三菱制药获得全球开发权益的一种可以阻断血清素、sigma 和α肾上腺素能受体的药物,Roluperidone旨在避免直接阻断多巴胺能受体(第一代和第二代抗精神病药的关键药理学靶点),同时保持对5-HT2A 的特定亚型血清素受体(第二代抗精神病药的另一个关键靶点)以及其他药理学靶点(sigma2 和肾上腺素能-α1A)的阻断,用于治疗精神分裂症患者阴性症状[4]。
精神分裂症是一种慢性、严重和使人衰弱的精神疾病,其特征是思维、感知、情绪、语言、自我意识和行为的扭曲。精神分裂症影响着全世界2000万人。
阴性症状会使精神分裂症患者变得对任何事情不感兴趣或无法完成任务或感到快乐。阴性症状的特征有五种结构:迟钝的情感、失语症、失语症、快感缺乏和不合群。
阴性症状是精神分裂症患者功能不良的主要原因,也可能是超高危青少年可能发展为全面精神分裂症的主要原因之一。目前,美国尚无批准治疗精神分裂症阴性症状的治疗方法。
2022年8月份,Minerva首次向FDA提交Roluperidone用于治疗精神分裂症患者阴性症状的NDA。
此次NDA申请得到了两项针对中度至重度精神分裂症阴性症状和稳定阳性症状患者的后期对照研究的结果(研究MIN-101C03和MIN-101C07)的支持[5]。
MIN-101C03 研究结果显示:治疗 12 周后,Roluperidone在减轻精神分裂症的阴性症状方面优于安慰剂。在主要疗效分析中,根据PANSS 五角结构模型阴性评分(PSM)测量(p ≤ 0.0036),64 mg Roluperidone导致精神分裂症阴性症状在统计学上显着减少。
对PANSS Marder阴性症状因子评分(NSFS)从基线到第 12 周的变化的事后分析也表明,与安慰剂相比,64 mg Roluperidone组具有统计学意义差异 (p ≤ 0.001)(图2)。
在双盲(DB)期 12 周后,与安慰剂相比,64 mg Roluperidone在多项次要/探索性疗效分析中也观察到统计学上的显着改善。在 24 周开放标签(OL)期间,NSFS 也得到了进一步的改善。MIN-101C07研究也证明了Roluperidone优于安慰剂[6]。
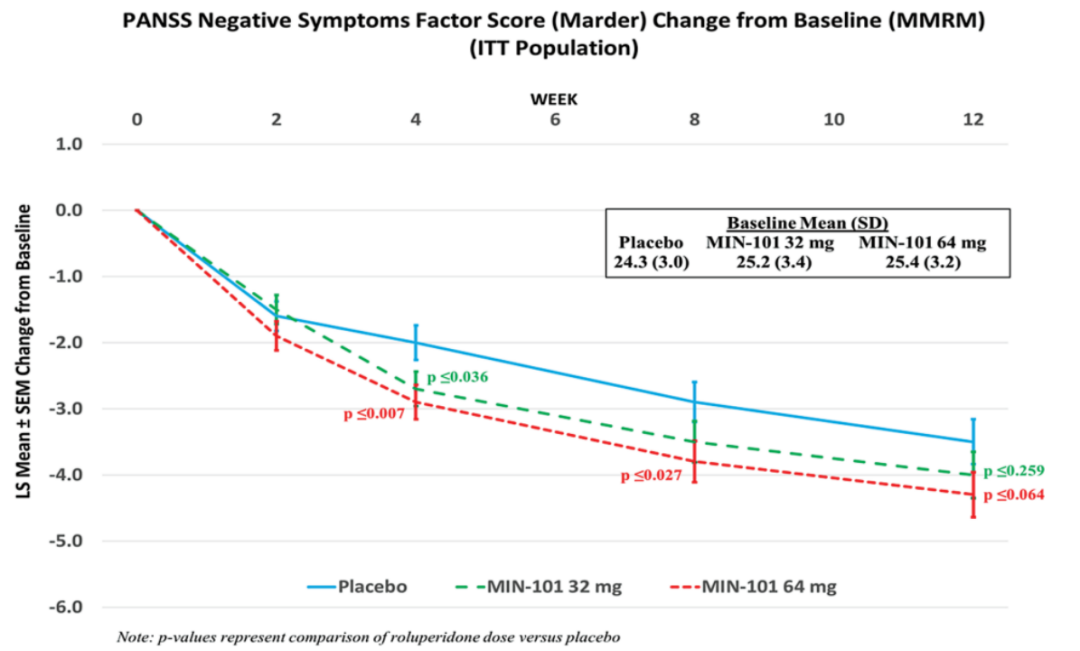
图2. Roluperidone治疗效果
然而2022年10月份,Minerva表示公司收到FDA拒绝Roluperidone的NDA。FDA表示,Minerva可以要求召开A类会议,讨论拒绝提交信函的内容[7]。
2023年5月10日,Minerva更新了Roluperidone的NDA近况,表示FDA于2023年5月8日重新接受了Roluperidone的NDA,FDA 已将PDUFA的目标日期定为2024年2月26日[9]。如果获得批准,Roluperidone可能是一个重要的新选择,可以解决精神分裂症患者群体面临的严重未满足需求,这些患者的阴性症状是残疾的主要来源,并对他们的日常生活质量产生不利影响。
04
Resmetirom
PDUFA日期:2024.03.14
Resmetirom是由Madrigal Pharmaceuticals研发的每日一次的口服甲状腺激素β 受体(THR-β)选择性激动剂,用于治疗非酒精性脂肪性肝炎(NASH)。
甲状腺激素通过激活肝细胞中的β受体,在影响一系列健康参数的肝功能中起着核心作用。
甲状腺激素信号转导障碍会加重肝脏脂毒性,导致非酒精性脂肪性肝病(NAFLD)进展为NASH并伴有肝纤维化。在人NASH中,肝脏具有低THR-β活性,加剧了线粒体功能障碍,脂肪毒性和纤维化。
研究发现THR-β激动剂显示出减少炎症性肝脂肪和纤维化的潜力,同时降低胆固醇和其他致动脉粥样硬化脂质。
Madrigal目前正在进行四项3期临床试验,以评估Resmetirom治疗NASH的安全性和有效性:MAESTRO-NASH、MAESTRO-NAFLD-1、MAESTRO-NAFLD-OLE 和 MAESTRO-NASH-OUTCOMES[8]。
2023年4月18日,Madrigal宣布Resmetirom获得FDA的突破性疗法认定,用于治疗伴有肝纤维化的NASH患者。公司还宣布,3期MAESTRO-NASH活检试验已完成入组[9]。
Madrigal在2023年7月完成向美国FDA滚动提交Resmetirom的NDA,用于治疗伴肝纤维化的NASH成人患者,2023年9月13日,Madrigal宣布FDA已接受Resmetirom用于治疗患有肝纤维化的成年NASH患者的NDA请审查。FDA已授予优先审查, PDUFA是2024年3月14日[10]。
2023年11月10日,Madrigal Pharmaceuticals公布了resmetirom的3期MAESTRO-NASH试验的新数据:绝大多数(>70%)接受resmetirom 100mg治疗的患者的MRI-PDFF减少了≥30%,达到了试验的主要肝活检终点和降低低密度脂蛋白胆固醇的关键次要终点,证明了Resmetirom对肝脏健康非侵入性措施的广泛治疗效果[11]。
05
Sotatercept
PDUFA日期:2024.03.26
Sotatercept是由Acceleron公司原研的一款潜在“First-in-class“的IIA型激活素受体(ACVR2A)融合蛋白,由人Activin受体IIA的胞外结构域与IgG1的Fc结构域融合而成,阻断激活素与细胞膜上的受体结合,从而降低激活素介导的信号传导。
2021年9月,默沙东以115亿美元收购Acceleron,获得了新批准上市的贫血药物Reblozyl和在研肺动脉高压药物sotatercept。
早在2020年4月份,Acceleron获得FDA突破性疗法认定,用于治疗肺动脉高压(PAH),成为首 款获得突破性疗法认定的PAH在研疗法[12]。
2023年8月1日,默沙东在发布2023年上半年的财务报告时表示,已向FDA递交Sotatercept用于治疗PAH生物制品许可申请(BLA)。
PAH是一种罕见的、进行性的、危及生命的血管疾病,其特征是肺小动脉收缩和肺循环血压升高。美国约有 40,000人患有 PAH。对于许多患者来说,这种疾病进展迅速。PAH会对心脏造成严重压力,导致体力活动受限、心力衰竭和预期寿命缩短。PAH患者的5年死亡率约为43%。
2023年9月28日,默沙东公司宣布FDA已接受Sotatercept用于治疗PAH成人患者的BLA的优先审评,FDA 将PDUFA定为 2024年3月26日[13]。
此次优先审评基于 3 期 STELLAR 试验的数据:sotatercept 在在6分钟步行距离(6MWD)方面和9项次要结局指标中的8项显示出具有统计学意义和临床意义的改善(图3)[14]。
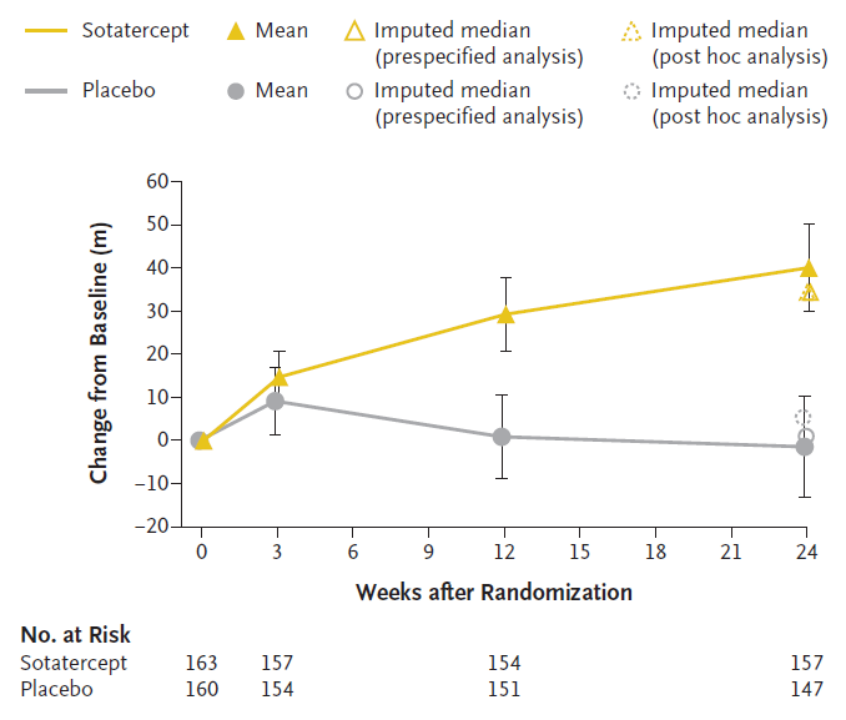
图3. 第24周6 钟步行距离的变化
06
Vadadustat
PDUFA日期:2024.03.27
Vadadustat是由大冢制药研发的一种口服缺氧诱导因子脯氨酰羟化酶(HIF-PH)抑制剂,已于2020年6月在日本获得批准,用于成人患者治疗慢性肾脏病(CKD)相关贫血。
HIF-PHI 旨在模拟人体对较低氧气水平的反应,例如当一个人处于高海拔地区时身体通过增加HIF的可用性来自然地对较低的氧气水平做出反应,HIF是一种协调促红细胞生成素合成和调节铁代谢的基因表达的蛋白质。
抑制HIF-PH可导致红细胞生成增加并改善向组织的氧气输送。HIF的发现为帮助理解氧传感在许多疾病中的核心作用奠定了基础,包括CKD引起的贫血。
早在2021年,新英格兰医学杂志上发布了关于Vadadustat治疗慢性肾脏病(CKD)和透析患者肾性贫血的研究结果:在两项针对透析依赖性慢性肾脏病(INNO2VATE)患者的随机(1:1)、全球、3 期、开放标签、申办者盲法、平行组、主动对照非劣效性试验中,vadadustat 在心血管安全性和血液学疗效方面不劣于达依泊汀α。
2023年9月29日,Sarnak 等人在肾病透析移植(NDT)杂志发布了Vadadustat相关研究的最新进展。
研究结果显示:在接受腹膜透析的患者中,初次评估期间血红蛋白浓度平均变化的差异为 -0.10 g/dL(95% CI -0.33,0.12)。Vadadustat和达依泊汀α组治疗中出现的不良事件的发生率(TEAE)分别为88.2%和95.5%,严重TEAE 分别为52.6%和73.2%(图4)[15]。
研究结果表明在3期INNO2VATE试验中接受腹膜透析的患者亚组中,vadadustat的安全性和有效性与达依泊汀α相似。
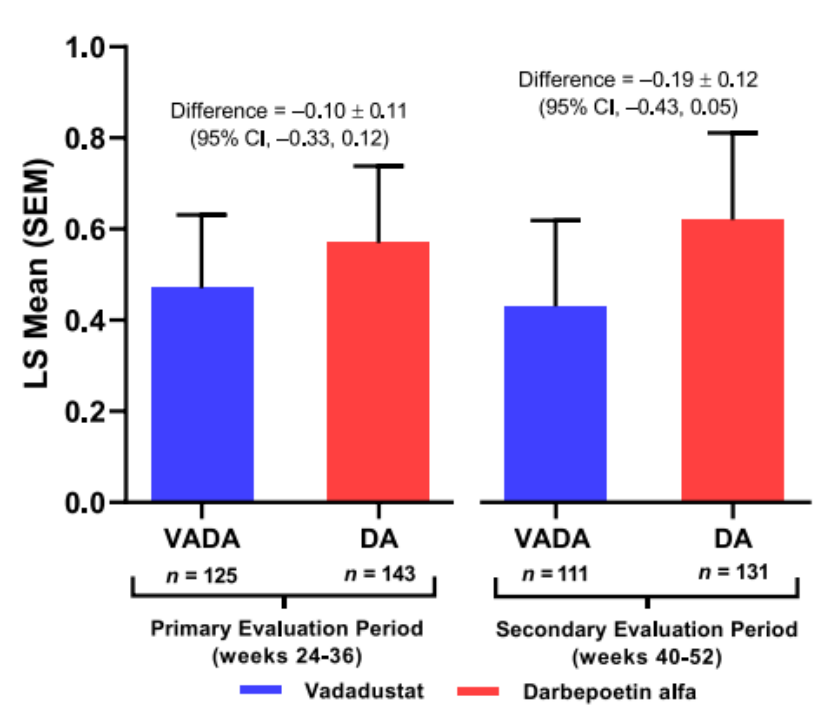
图4. 3期INNO2VATE试验结果
2021年3月,Akebia向FDA提交了Vadadustat的NDA,然而2022年3月29日,FDA拒绝了Vadadustat的NDA。
2023年10月份,Akebia 向FDA重新提交了Vadadustat的NDA,用于治疗接受透析的成年患者CKD引起的贫血,FDA 将PDUFA定为2024年3月27 日[16]。
07
Donanemab
Donanemab是由礼来研发的一款Ab单抗药物,用于治疗阿尔兹海默症。阿尔兹海默症发病机制不明,其中Aβ的生成、清除失衡和Tau蛋白过度磷酸化导致炎症反应、神经元死亡等一系列病理过程是目前比较公认的AD发病机制。
2023年7月6日,卫材/Biogen的Lecanemab获得了FDA完全批准,成为了20年来首 款获得FDA完全批准的AD新疗法,开启了其真正的商业化道路。
相对于Lecanemab,Donanemab的优势是注射间隔更长,每四周注射一次,Lecanemab是两周一次注射,患者负担更大,是显著劣势。
2022年,礼来向FDA提交了Donanemab的NDA,然而2023年1月19日, FDA拒绝了donanemab用于治疗早期症状性阿尔茨海默病的加速批准,原因是提交材料中提供的药物暴露数据至少12个月的患者数量有限。没有发现申请中的其他缺陷。
随后2023年7月份,礼来公布了donanemab在3期TRAILBLAZER-ALZ 2临床试验中取得积极结果:1. Donanemab显著减缓了淀粉样蛋白阳性早期症状性阿尔茨海默病患者的认知和功能下降,降低了疾病进展的风险;2. 近一半接受Donanemab治疗的疾病早期受试者在1年时没有临床进展;3. 其他亚群分析显示,那些处于疾病早期阶段的研究参与者有更大的益处,与安慰剂相比,下降速度减缓了60%;4.在试验过程中,尽管许多受试者在6个月或12个月时完成了疗程,但相对于安慰剂,治疗效果继续增加,支持有限时间的给药[17]。
礼来将该试验结果作为补充材料继续向FDA提交了Donanemab的NDA,2024年Donanemab有望获FDA批准上市。
2023年10月31日,Donanemab在国内的上市申请已被CDE正式接受。此前,NMPA对Donanemab给予了突破性治疗药物认定,用于治疗早期阿尔茨海默病,包括阿尔茨海默病引发的轻度认知障碍和轻度阿尔茨海默病。
08
Tovorafenib
PDUFA日期:2024.04.30
Tovorafenib是由Day One Biopharmaceuticals公司开发的一种口服、脑穿透性pan-RAF激酶抑制剂,能够抑制野生型和某些突变形式的BRAF、CRAF和ARAF蛋白激酶。
2023年10月30日,Day One宣布FDA接受Tovorafenib用于复发或进展性小儿低级别胶质瘤(pLGG)的新药上市申请并获得优先审评,PDUFA目标行动日期为2024年4月30日。
该NDA基于一项开放标签的关键性2期试验的结果,该试验评估Tovorafenib作为6个月至25岁复发或进展性pLGG患者每周一次的单药治疗。
2023年11月,Tovorafenib的这项临床2期(FIREFLY-1)试验结果被发表在Nature Medicine 上,研究结果显示Tovorafenib可能是治疗携带BRAF突变的复发/难治性儿童低级别胶质瘤的有效疗法,为治疗儿童低级别胶质瘤带来新希望 [18]。
研究结果显示:根据独立审查评估,在神经肿瘤学高级别胶质瘤(RANO-HGG)患者中,总体缓解率(ORR)为67%,达到第1组预先设定的主要终点,中位缓解持续时间(DOR)为 16.6 个月,和中位时间缓解(TTR)为3.0个月(次要终点)(图5)。
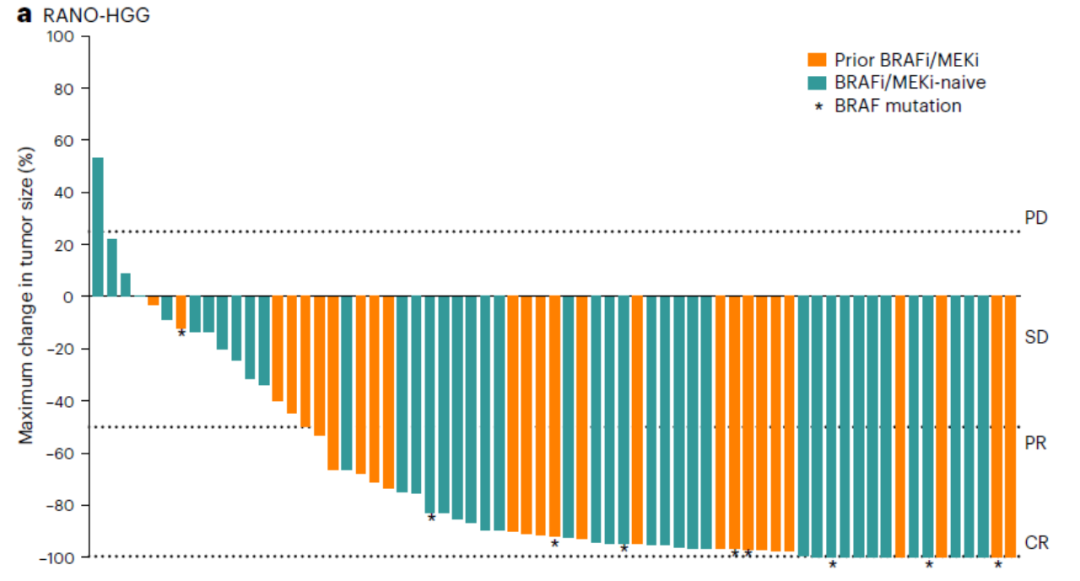
图5. RANO-HGG患者中疗效
在小儿神经肿瘤学低级别胶质瘤(RAPNO)患者中,ORR为51%,中位DOR为13.8个月,中位TTR为5.3个月(图6)。
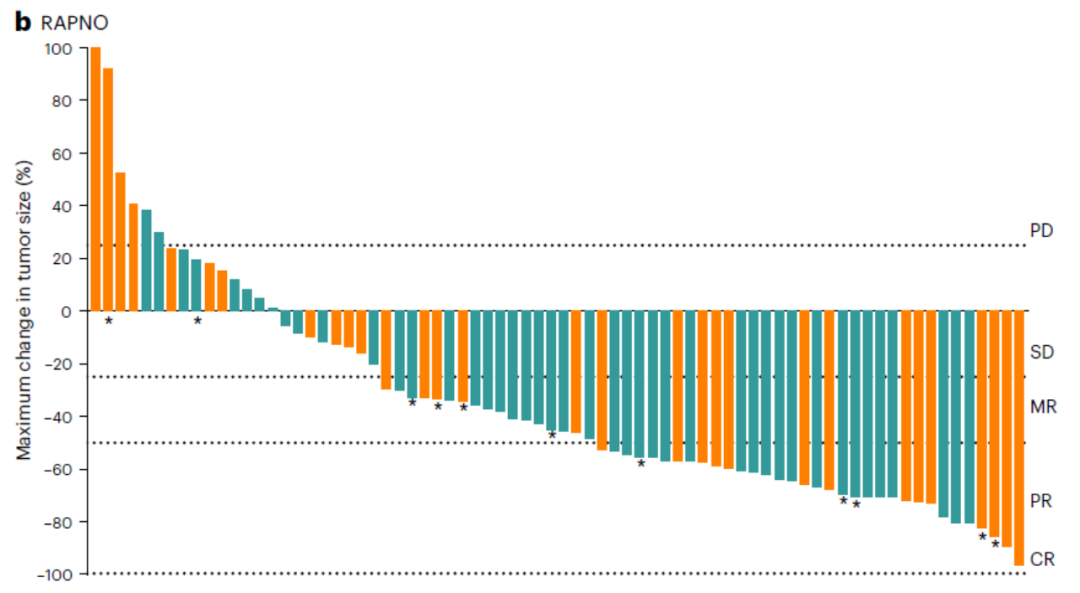
图6. RAPNO患者中疗效
09
Imetelstat
PDUFA日期:2024.06.16
Imetelstat是由Geron Corporation研发的“First-in-Class”的端粒酶抑制剂, 用于治疗低危骨髓增生异常综合征(MDS)患者的输血依赖性贫血。
2023年6月份,Geron 向FDA提交了Imetelstat的NDA,随后8月21日,Geron宣布FDA已接受Imetelstat的NDA,将PDUFA日期定为2024年6月16日。
此次NDA基于3期IMerge试验的结果,其中imetelstat组的8周输血独立性(TI)的主要终点显著高于安慰剂组(p<0.001),imetelstat 8周TI应答者的中位TI持续时间接近一年,与安慰剂患者相比,imetelstat治疗患者的平均血红蛋白水平随时间显着增加(p<0.001)[19]。
Bruedigam等人在Nature Cancer上报道了imetelstat在一项针对患者来源的异种移植物的随机 II 期样临床前试验结果,imetelstat 可有效减轻急性髓系白血病(AML)负荷,并优先靶向含有突变 NRAS 和氧化应激相关基因表达特征的亚组[20]。
10
Patritumab deruxtecan
PDUFA日期:2024.06.26
Patritumab deruxtecan(HER3-DXd)是由第一三共原研的一款靶向HER3的ADC药物,通过马来酰亚胺-GGFG接头将HER3单抗patritumab与拓扑异构酶I抑制剂deruxtecan连接而得(图7)。
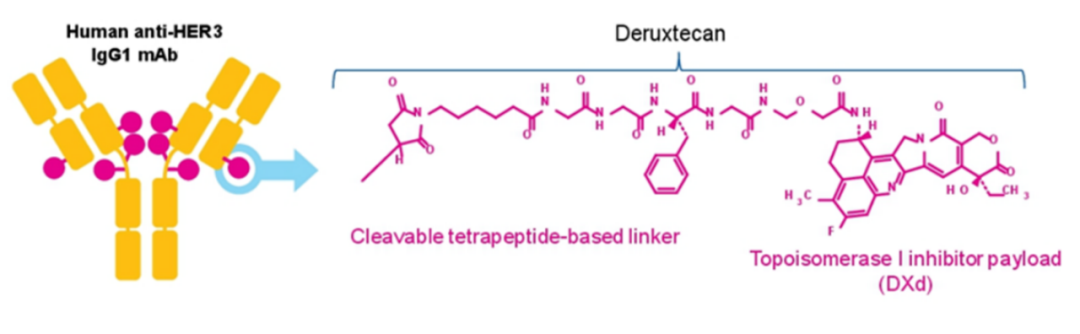
图7. Patritumab deruxtecan的结构
2023年10月20日,默沙东与第一三共就包括patritumab deruxtecan在内的三款ADC药物达成了高达220亿美元的合作协议,共同开发和商业化这些产品。
2023年12月22日,第一三共和默沙东共同宣布patritumab deruxtecan的BLA获FDA受理并予以优先审评,适应症为既往至少接受过两种系统治疗的EGFR突变型局部晚期或转移性非小细胞肺癌(NSCLC),成为首 款申报上市的HER3 ADC,PDUFA日期为2024年6月26日。
BLA基于HERTHENA-Lung01关键性2期试验的主要结果和IASLC 2023世界肺癌大会(#WCLC23)上公布的数据结果,这些结果同时发表在《临床肿瘤学杂志》上。
HERTHENA-Lung01研究结果显示:在EGFR TKI和铂类化疗后疾病进展的225例EGFR突变局部晚期或转移性NSCLC患者中patritumab deruxtecan治疗的客观缓解率(ORR)为29.8%(95%CI:23.9-36.2),包括1例完全缓解和66例部分缓解。中位缓解持续时间为6.4个月(95%CI:4.9-7.8)[21]。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























