2024年8月19日,宜联生物和BioNTech同时发布声明,宣布已于2024年8月15日获FDA通知正式解除了对YL202/BNT326临床I期试验(NCT05653752)的Partial Hold。
2024年8月19日,宜联生物和BioNTech同时发布声明,宣布已于2024年8月15日获FDA通知正式解除了对YL202/BNT326临床I期试验(NCT05653752)的Partial Hold。



宜联生物和BioNTech发布声明
YL202/BNT326是一款靶向HER3的在研ADC药物,目前由宜联生物和BioNTech合作进行开发。该临床试验将会继续受试者招募,并将集中在表现出优异有效性及可控安全性的3.0 mg/kg及以下的剂量水平进行开发。
2023年10月,宜联生物YL202与BioNTech达成海外合作授权(BioNTech内代号BNT326),BioNTech向宜联生物支付7000万美元首付款及潜在超10亿美元总付款,获得YL202海外开发、制造和商业化的独家权利。
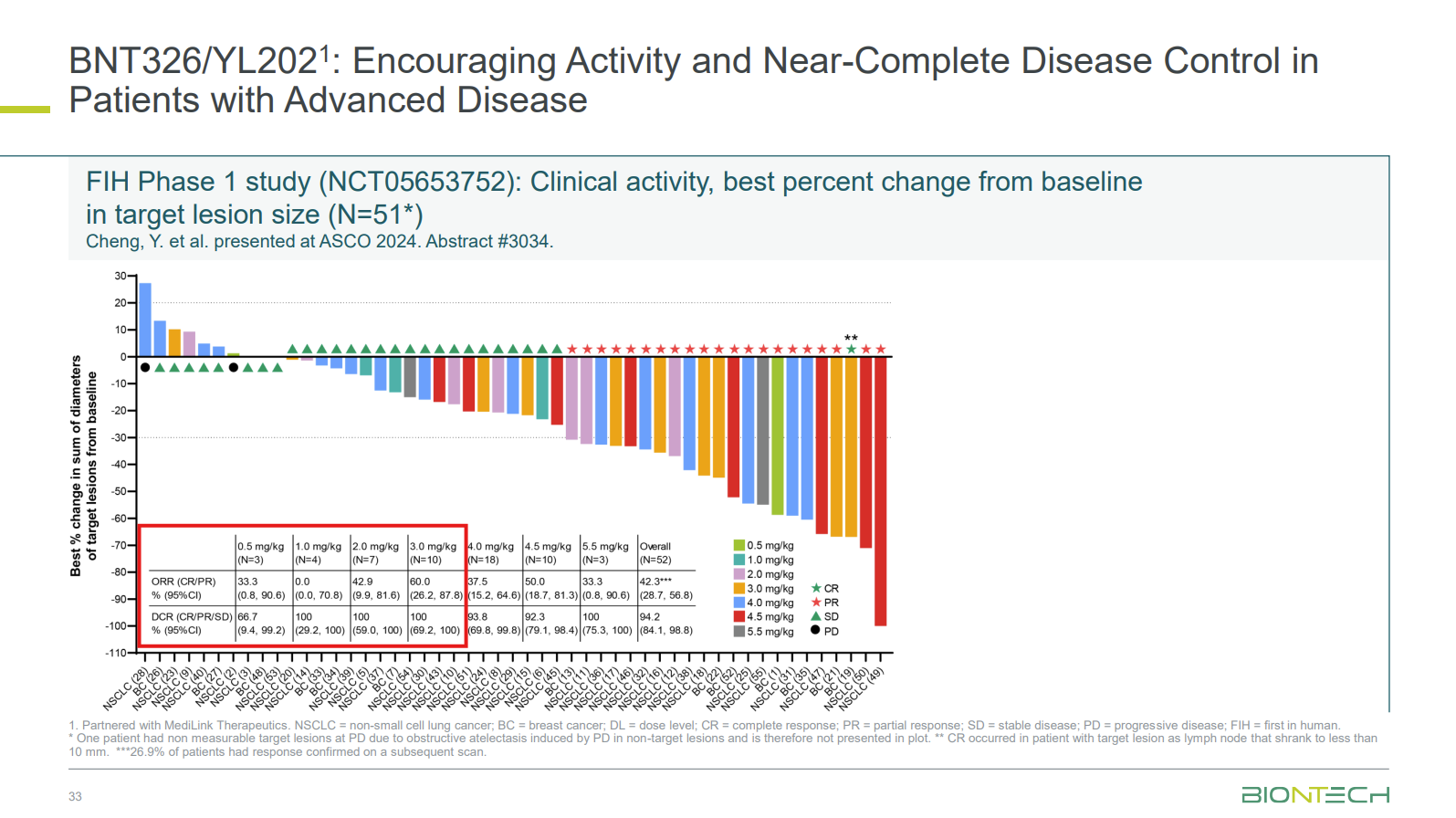
在ASCO 2024年会上,宜联生物公布HER3-ADC YL202-INT-01-01试验的初步积极结果,共入组52例患者,其中39例为接受过3代TKI和含铂化疗的携带EGFR激活突变的局部晚期或转移性非小细胞肺癌(NSCLC)患者和13例接受CDK4/6抑制剂和至少一线化疗的不可切除的、局部晚期或转移性HR+/HER2-乳腺癌(BC)患者。
YL202-INT-01-01试验的初步结果
在46例可评估药效患者中,ORR为37.0%(17/46 pts),DCR为93.5%;在DL3至DL5剂量组中,ORR为41.0%(16/39 pts),DCR为94.9%;在乳腺癌患者中ORR为54.5%(6/11 pts),DCR为100%。在NSCLC患者中的ORR可能只有36%。
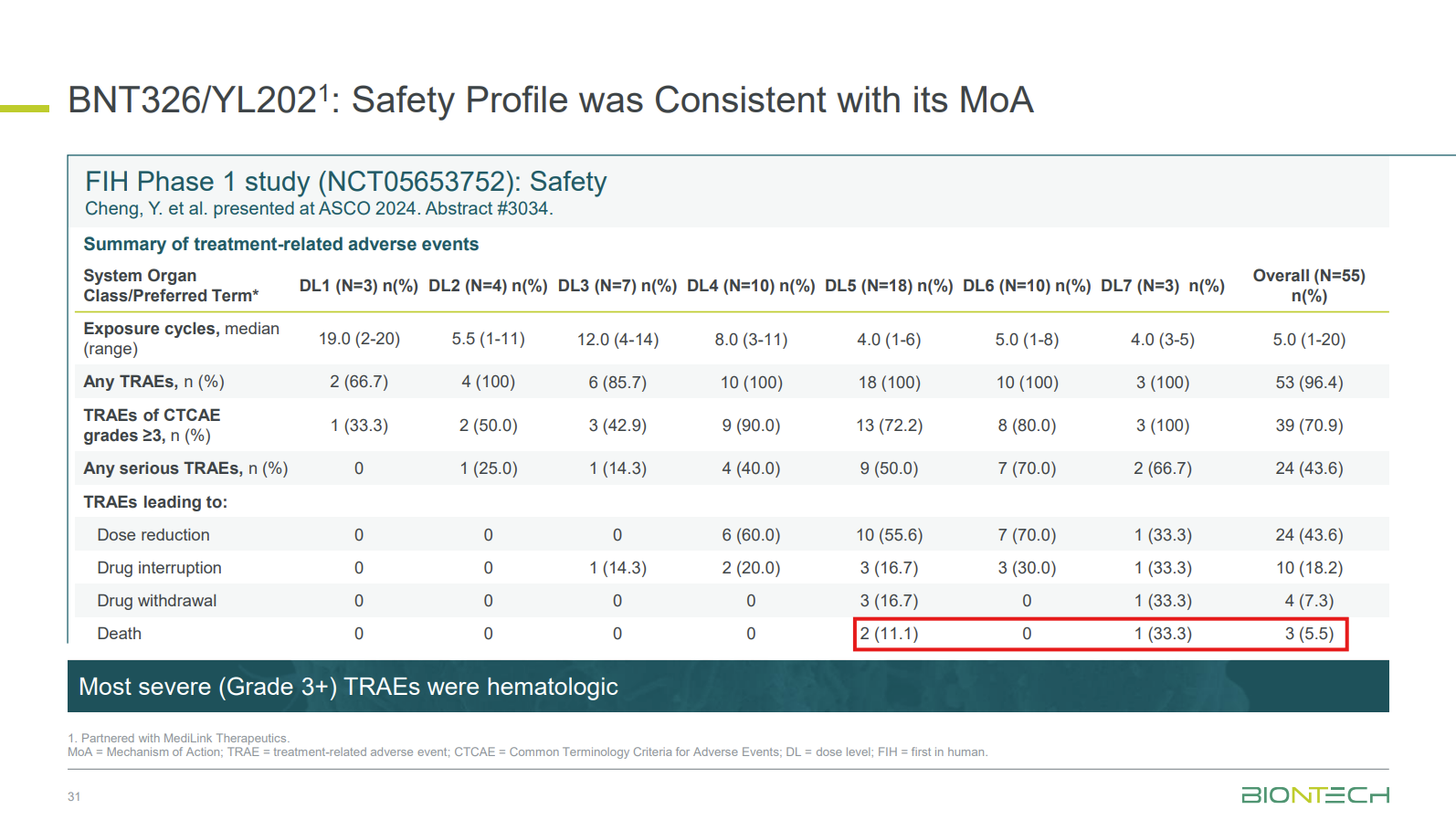
然而在2024年6月17日,FDA要求部分暂停正在进行的BNT326/YL202的I期试验(NCT05653752),暂停新受试者入组。原因是,BNT326/YL202可能在高剂量时造成“不可接受且显著的疾病或伤害风险”。FDA要求审查临床和安全数据,与该机构共享可用的药理学数据,并在研究者手册中提供有关安全发现的额外信息,包括在研究YL202-INT-101-01和YL202-CN-201-01中观察到的5级不良事件(3例)。
YL202-INT-01-01安全性结果
4mg/kg剂量组,2例患者在发热性嗜中性粒细胞减少和全血细胞减少后因败血症致死;5.5mg/kg剂量组中1例患者感染新冠后得间质性肺炎死亡。
当然,BioNTech也观察到BNT326/YL202治疗相关不良事件呈剂量依赖性趋势,特别是中性粒细胞计数减少和粘膜炎事件发生率增加这一结果,并在ASCO幻灯片中表示,“进一步的临床开发将集中在4.0 mg/kg以下的剂量水平,该剂量水平的安全性是可控的,并且观察到有希望的临床活性。”
YL202-INT-01-01疗效结果
通过对结果的进一步分析来看,3.0mg/kg及以下剂量的耐受性较好,并且有效性仍然能够保证。
如今,FDA解除对BNT326/YL202的临床暂停,宜联生物和BioNTech将进一步优化临床试验,BioNTech补充道:“根据正在进行的BNT326/YL202 评估试验中新出现的安全性数据,公司迅速而主动地采取了预防措施,包括不再招募剂量高于3mg/kg的患者,并降低试验中已招募较高剂量水平的参与者的剂量水平。宜联生物与BioNTech合作分析新出现的数据并实施进一步的风险缓解措施,包括数据分析、更新研究者手册、患者知情同意书和临床试验方案,并修订关于剂量延迟、减少和修改以及针对 TRAE 的预防性药物的指导,以符合FDA的要求。”
YL202的2期临床
并且宜联生物目前仍在积极推进YL202,根据clinical trails网站查询,宜联医药登记了一项HER3 ADC药物YL202,在TNBC、HR 阳性、HER2零表达或HER2低表达的局部晚期或转移性乳腺癌患者的疗效、安全性和药代动力学的2期临床试验NCT06439771。
资料来源:
2.FDA Lifts Partial Hold on BioNTech, MediLink’s ADC Cancer Trial.August 19, 2024.Tyler Patchen
3.FDA allows BioNTech-MediLink ADC trial to resume with lower, safer doses.By James Waldron Aug 19, 2024





 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























