一、生物药的特性与挑战
生物药(如单克隆抗体、生长因子等)具有复杂的分子结构和生物活性,其生产依赖于生物技术手段(图 1)。与化学药相比,生物药的制造过程更易受生物系统自然变异性影响,导致批次间存在微小异质性(图 2)。欧盟通过严格的 GMP 标准和持续工艺验证,确保生物药的质量一致性。
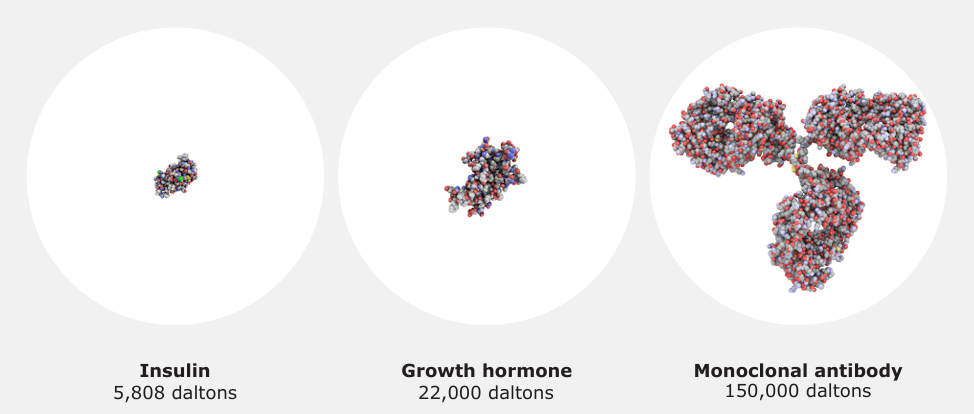
图 1:欧盟批准生物药的蛋白质类型示例(来源:EMA, 2019)
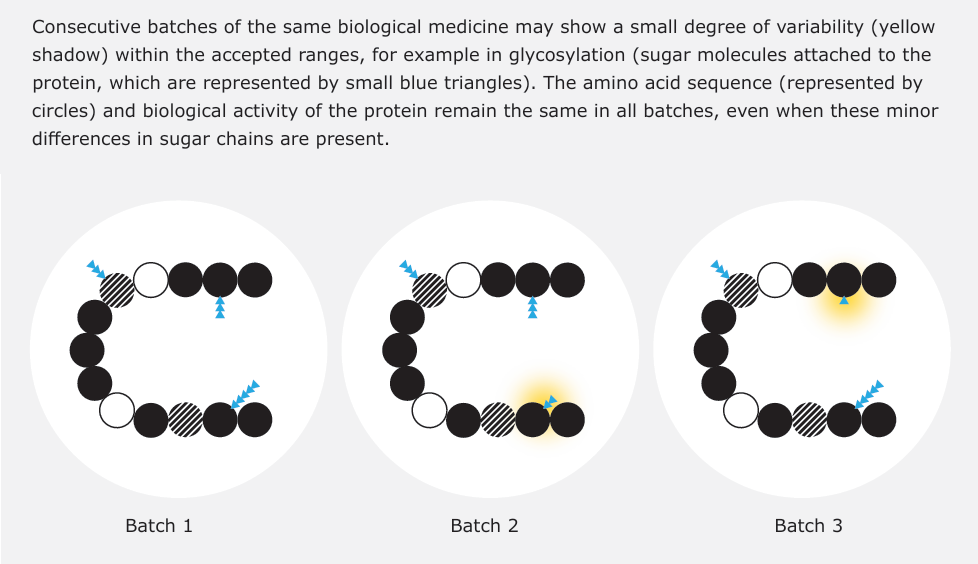
二、生物类似药的定义与开发路径
生物类似药是与已上市参照药高度相似的生物制品,允许存在不影响临床疗效和安全性的微小差异(表 1)。其开发需通过三步比较研究:
1.质量对比:利用质谱、细胞活性分析等技术验证分子结构和功能一致性
2.非临床研究:通过体外/体内实验评估药效学和毒理学特征
3.临床研究:采用等效性设计验证药代动力学、免疫原性及疗效(图 5)
表 1:生物类似药的核心特征(来源:EMA, 2019)
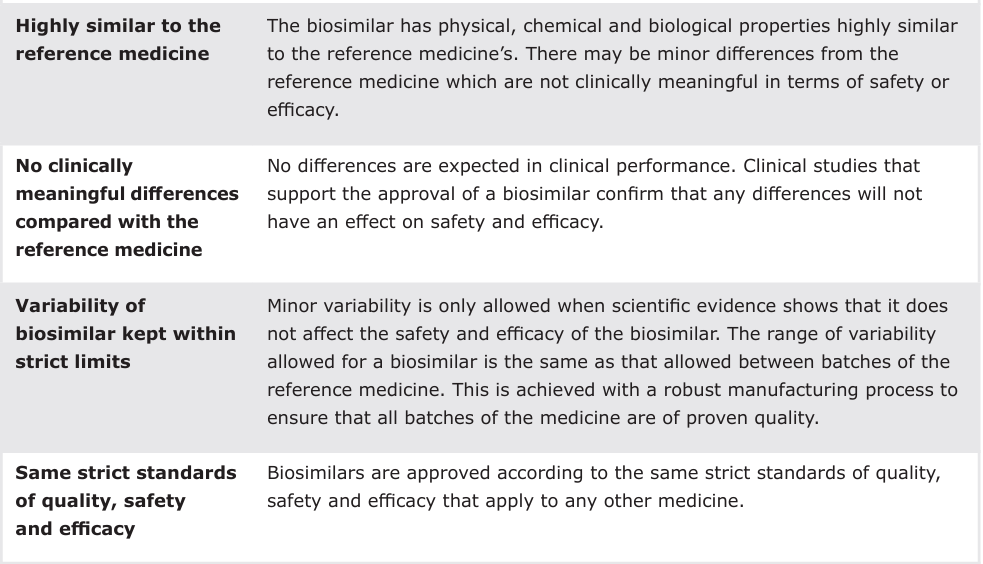
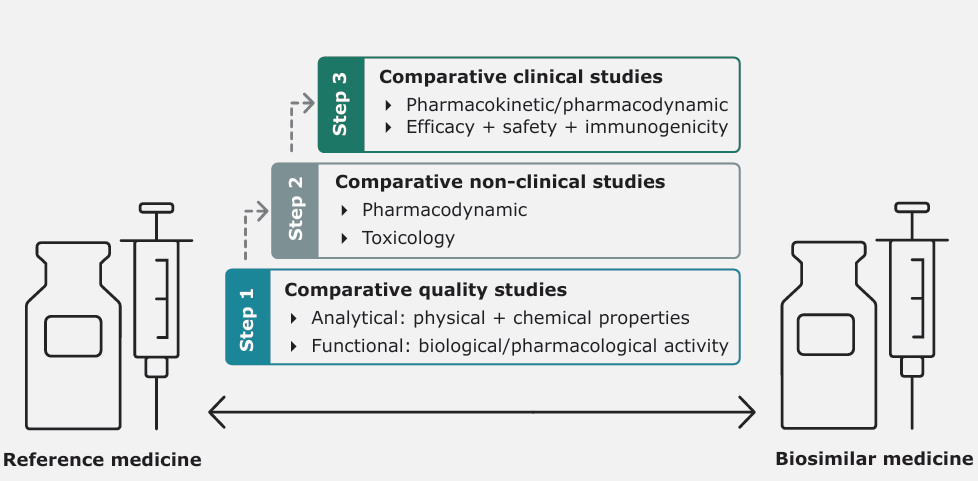
图 5:生物类似药开发的三步对比研究(来源:EMA, 2019)
三、审批标准与数据要求
欧盟采用 "逐步对比" 策略,通过质量研究数据减少临床研究需求。对于复杂分子(如单抗),需开展患者等效性试验;而简单分子(如重组人粒细胞集落刺激因子)可仅通过健康志愿者药代动力学研究获批。数据外推机制允许将参照药适应症扩展至生物类似药,前提是作用机制和安全性特征具有一致性(表 5)。
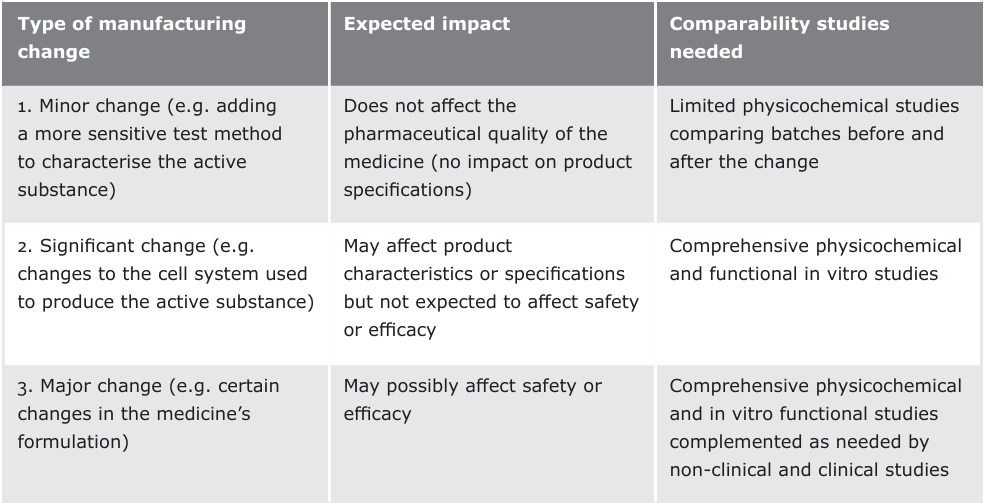
表5:生物技术生产药物制造过程变更后需要的可比性研究(来源:EMA, 2019)
四、全生命周期安全管理
欧盟建立了与参照药同等严格的安全监测体系:
● 风险管理计划(RMP):包含上市后安全性研究(PASS)和风险最小化措施
● 额外监测:新批准生物药需标注黑三角标识,强化不良反应报告
● 追溯系统:要求记录商品名和批次号,确保供应链全程可追溯
数据显示,2006-2019 年间欧盟批准的 50 余种生物类似药中,未因安全性问题撤市,其不良反应发生率与参照药无显著差异。
五、互换性与市场影响
欧盟明确互换性决策属于成员国权限,但要求医生在换药时需考虑患者个体情况。生物类似药竞争使欧洲医疗系统节省约 30% 的生物药支出,同时促进创新疗法可及性。例如,2018 年生物类似药占欧盟生物药市场份额的 15%,预计 2025 年将增至 30%。
六、全球监管领导力
欧盟的生物类似药标准被 WHO 和美国 FDA 等国际机构广泛采纳。EMA 通过国际协作推动生物类似药开发的科学共识,其指南文件(如 CHMP/BMWP/42832/2005)成为全球监管参照基准。
结论
欧盟的生物类似药监管体系通过科学严谨的对比研究、动态安全监测和透明的信息公开,在保障患者安全的同时促进医疗资源优化。医药专业人员应基于 EPAR 评估报告(含详细对比数据)和 SmPC 规范用药,共同推动生物类似药的合理应用。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























