1、杂质之关键知识点
杂质定义
按照《化学药物杂质研究技术指导原则》定义,原料药中所含的杂质是指任何影响药物纯度的物质,是药物中存在的无治疗作用、或影响药物的稳定性和疗效、甚至对人体健康有害的物质,从其化学结构来分析是与原料药存在差异的某种成分。简言之,它是存在于某一新原料药中任何影响药品纯度的物质。
杂质的分类
按其理化性质一般分3类:
有机杂质(主要为工艺杂质、降解杂质、有关物质)、无机杂质(反应试剂、配位体、催化剂、重金属、其它残留的金属、无机盐、助滤剂、活性炭等)及残留溶剂(原料药及制剂生产过程中使用的有机溶剂)。
按照其来源,杂质可以分为工艺杂质(包括合成中未反应完全的反应物及试剂、中间体、副产物等)、降解产物、从反应物及试剂中混入的杂质等。
按照其**分类,杂质又可分为**杂质和普通杂质等。
杂质分析方法及验证
分析方法的选择直接关系到杂质测定结果的专属性与准确性,因此,在进行杂质研究时首要问题是选择合适的杂质分析方法。
而方法验证应参照相关的技术指导原则进行,重点在于专属性和灵敏度的验证。专属性系指在其它成分可能共存的情况下,采用的方法能准确测定出被测杂质的特性。检测限是反映分析方法灵敏度的一个重要指标,所用分析方法的检测限一定要符合质量标准中对杂质限度的要求,检测限不得大于该杂质的报告限度。
PS:强降解试验对于未知杂质的分离度考察是非常必要的,其目的主要是提供关于杂质(特别是降解物)与主成分的分离情况、样品稳定性及降解途径等重要信息,考察此情况下的分离度更具有实际意义。
杂质限度的制订
在制订质量标准中杂质的限度时,安全性为首要考虑内容,尤其对于有药理活性或**的杂质;其次应考虑生产的可行性及批与批之间的正常波动;还要考虑药品本身的稳定性。
可根据稳定性数据、原料药的制备工艺、制剂工艺、降解途径等的研究及批次检测结果来预测正式生产时产品的杂质概况。当杂质有特殊的药理活性或**时,分析方法的定量限及检出限应与该杂质的控制限度相适应。设定的杂质限度不能高于安全性数据所能支持的水平,同时也要与生产的可行性及分析能力相一致。在确保产品安全的前提下,杂质限度的确定主要基于中试规模以上产品的实测情况,考虑到实际生产情况的误差及产品的稳定性,往往对限度做适当放宽。如果各批次间的杂质含量相差很大,则应以生产工艺稳定后的产品为依据,确定杂质限度。
创新药关于有机杂质的限度确定
由于在创新药物的研究过程中,需通过一系列的药理毒理及临床研究来验证该药品的安全有效性,而研究所用的样品本身会包含一定种类与数量的杂质,所以如果在这些研究中并未明显反映出与杂质有关的毒副作用,即使有些杂质的含量超出了原料药或制剂常规要求(见下表)的质控限度,仍可认为该杂质的含量已经通过了安全性的验证。
表1. 制剂的杂质限度
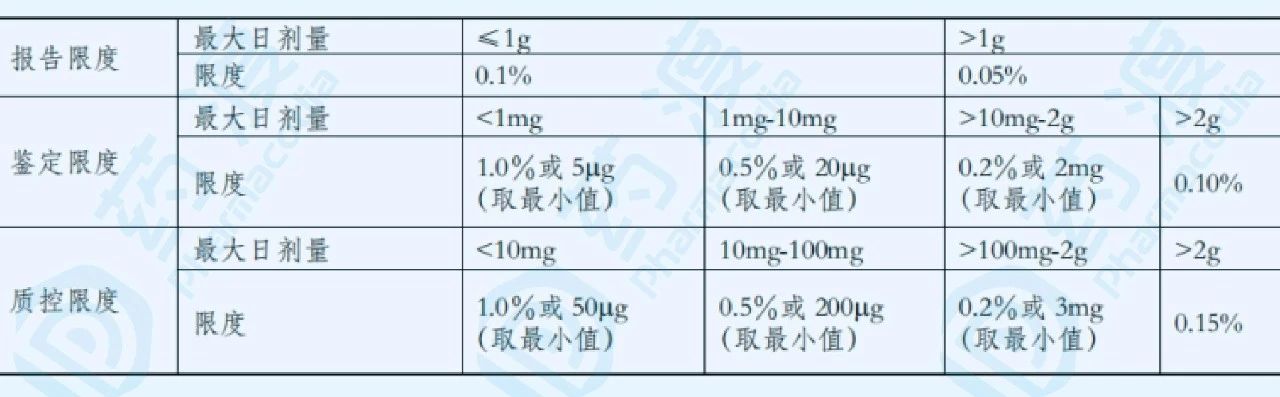
表2. 原料药的杂质限度
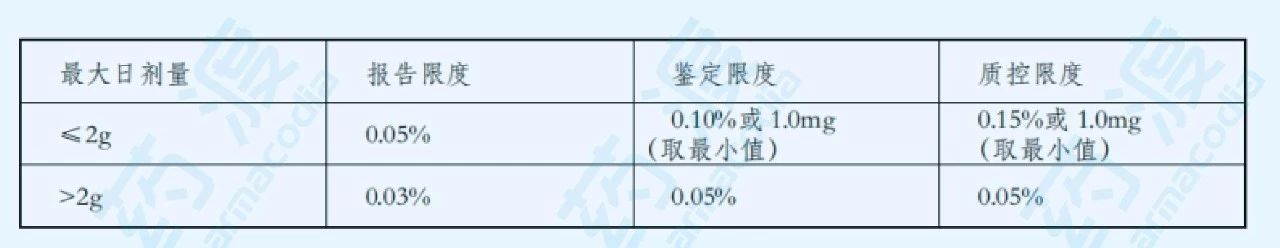
在此前提之下,如果该杂质的含量同时也在正常的制备工艺所允许的限度范围内,那么根据试验样品中杂质的含量所确定的限度可认为是合理的。同时,对上市后产品须进行新增不良反应的原因进行分析,如与杂质有关,则应分析原因,设法降低杂质含量,这样制订出来的杂质限度才能保证产品的安全性。如某杂质同时也是该药物在动物或人体中的主要代谢产物,则对该杂质可不考虑其安全性,但需制订合理的限度。对于用于某些适应症的药物,可以根据用药人群、剂量、用药周期、临床经验、利弊权衡等,对杂质的限度做适当的调整。当研究证明某些药物中的杂质与不良反应有关,则应在制订该杂质的限度时引起重视,并适当提高限度要求。反之,杂质的限度可适当放宽。杂质含量限度决策分析见下图。
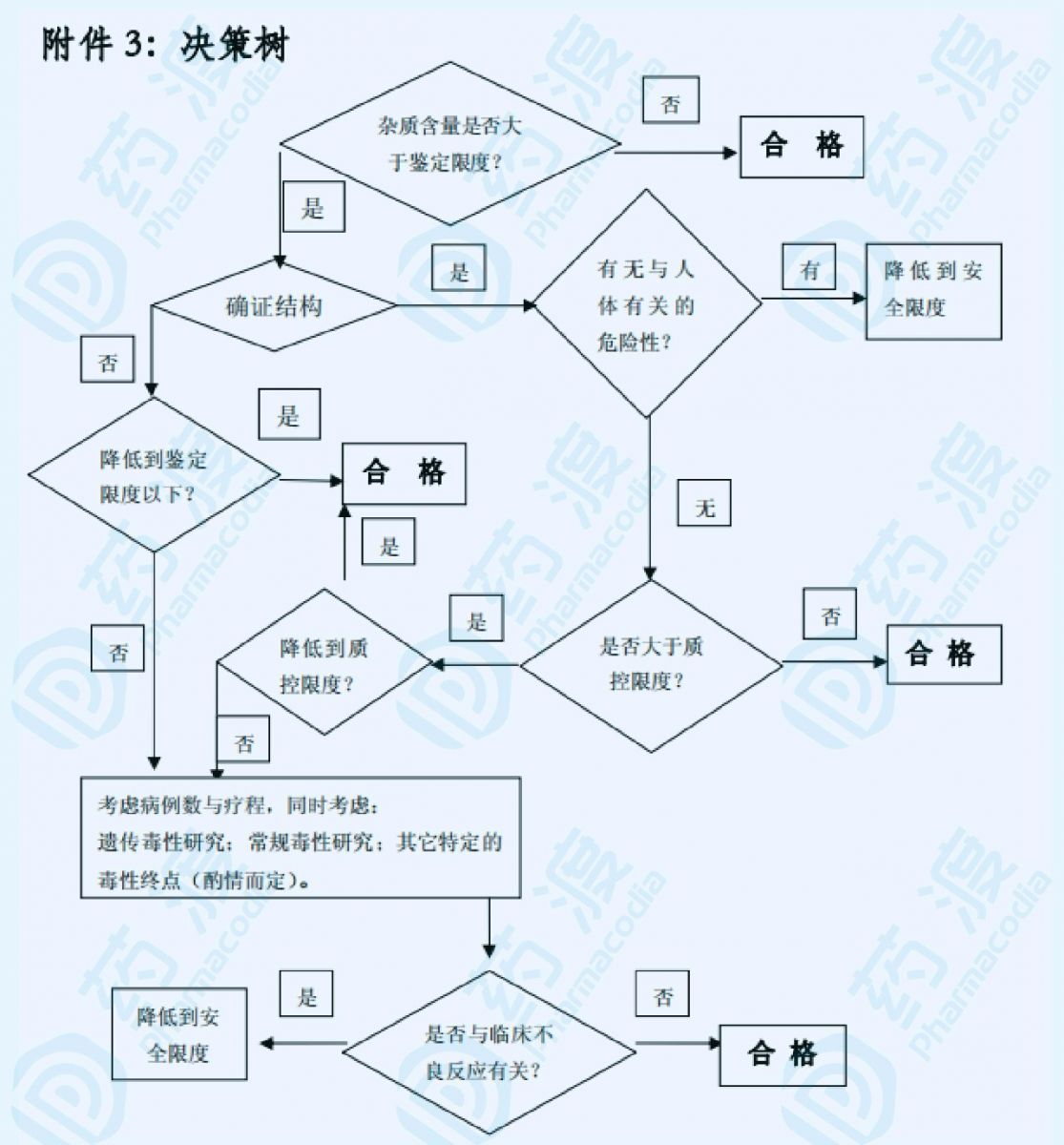
图1. 杂质限度分析决策树
临床研究申请与上市生产申请阶段的杂质研究
申临床时,杂质研究工作可从以下几方面考虑。1)安全性,在申报临床研究前,根据安全性研究用样品的杂质含量情况来证明临床研究用药品是安全的。2)同时,在临床研究期间对杂质分析方法进行完善。3)对于创新药物,杂质限度的最终确定需根据临床研究结果进行综合权衡。故在申报创新药物临床研究时,可对杂质的限度做一个初步的规定。
临床研究结束后,应将放大生产的样品与临床研究样品中的杂质进行详细比较,如因生产规模放大而产生了新的杂质,或已有杂质的含量超出原有的限度时,正常按照杂质限度进行决策分析即可。
2、杂质之知识点问与答
杂质研究应有的逻辑是什么?
基于杂质谱分析的杂质研究是一种“以源为始”的主动思维模式,以“质量源于设计”的观点,从杂质来源入手,从制备工艺、化学结构、处方组成的分析出发,评估、预测产品中可能存在的及潜在的副产物、中间体、降解物以及试剂、催化剂残留等大体的杂质概况,辅以适当的强制降解、对照物质的加入等验证的手段,考证建立的分析方法是否能够将它们逐一检出,并进行相应的方法学验证工作;相比之下,传统的杂质研究是一种“以终为始”的被动行为和逆向思维模式,从杂质分析的结果出发,仅从建立的某种检测方法所检出的有关物质中归属其来源情况,而未充分分析与验证可能存在的潜在杂质情况,建立的分析方法能否全面检出这些杂质,故容易出现杂质漏检的情况,难以全面掌握产品的杂质谱。
常说的“杂质谱”包含哪些内容?
杂质谱包括药物中所有杂质的种类、含量、来源及结构等信息。通过杂质谱分析较为全面地掌握产品中杂质概貌(种类、含量、来源和结构等);有针对性地选择合适的分析方法,以确保杂质的有效检出和确认;跟踪杂质谱对安全性试验或临床试验结果产生的影响,评估杂质的可接受水平;结合规模化生产时杂质谱的变化,评估杂质安全性风险,确立安全的杂质控制水平。
杂质分析方法的验证应具备怎样的基本条件?
简而言之为,针对性和全面性!杂质的微量性和复杂性,使得检测方法的专属性、灵敏度和准确度十分关键。杂质分析方法的对象是各个潜在的杂质,因此,其分析方法的验证需要根据不同杂质的特点综合设计验证方案,进行有针对性的规范验证。
杂质控制仅在“质量控制”模块吗?
CTD格式中杂质控制的考虑要体现在CMC的各个环节,而不是仅仅局限在“质量控制”模块,如制剂学科的原辅料控制。实际上这正是源头控制、过程控制与终点控制相结合的杂质控制理念的体现,在研发工作及申报资料的整理中都需要针对性的贯彻实施。
杂质研究中的“对比研究”是什么?
对比研究是桥接上市药物安全有效性信息的基础,获取上市产品杂质信息的重要途径,而且通过“类比思维”还可以减少研发工作的疏漏和复杂性。如BP收载了克林霉素磷酸酯的5种杂质的化学结构,**相差几个数量级,国内产品生产工艺与原研厂不同(无菌灌装),多经过了剧烈的终端灭菌过程,而主药水溶液高温不稳定,杂质总量与原研厂产品相比同样较高,但具体为何种杂质,**如何,与原发厂产品杂质是否相同,未进行此方面对比研究,很难桥接上市产品的安全性信息,加之未进行临床试验或动物安全性试验直接上市,其安全性风险极高。
创新药如何确定杂质限度?
创新药杂质限度的确定需要综合药学、药理毒理及临床研究结果综合判定,除了用含有一定量杂质的样品进行毒理试验外,也可以直接采用该杂质进行相应的毒理试验,为杂质限度的确定提供安全性方面的依据,这对于含可能具有遗传**结构单元的杂质是必须的,通过体外点突变和染色体畸变试验进行安全性考查,如果出现阳性结果,需通过优化工艺过程,消除其生成,否则,需在质量标准中严格控制。如果某已知杂质的**在相关的文献上已有详细记载,也可作为杂质限度确定的依据之一。
参考文献:
1. 国家食品药品监督管理局. 化学药物杂质研究的技术指导原则, 2005
2. 美国食品药品管理局. Guidance for Industry Drug Product Chemistry, Manufacturing and Controls Information [J/OL]. http://www.fda.gov/cder/guidance.
3.欧洲药品评价局. Guideline on the limits of genotoxic impurities[J/OL]. 2007. http://www.emea.eu.int.
4. 澳大利亚药品评价局. Guidelines for the Registration of Drugs, Vol.1, July 1994. Appendix 10.
5. 张哲峰, 鹿颐, 王元度, 梁贵键. RP-HPLC法分离测定洛伐他汀及其杂质.药物分析杂志. 1996(06)。
6. 高燕霞, 张哲峰, 鹿颐, 梁贵键. TLC及HPLC法分离测定克林霉素磷酸酯及其有关物质. 药物分析杂志, 1998(S1)。
7. 黄晓龙. 对创新药研发中杂质限度确定的几点思考. 中国新药杂志,2007(2)。
8. David Jacobson-Kram, TimothlyMcGovern Toxicological overview of impurities in pharmaceutical products, Advanced Drug Delivery Reviews, 2007(59): 38-42
9. CTD申报资料中杂质研究的几个问题. CDE官网



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























