paxalisib(GDC-0084)
近日,澳大利亚Kazia Therapeutics公司宣布,美国FDA已授予paxalisib(GDC-0084)治疗弥漫性内生性桥脑胶质瘤(Diffuse intrinsic pontine glioma, DIPG)的罕见儿科疾病资格(RPDD)。DIPG是一种罕见和高度侵袭性的儿童恶性肿瘤,极度缺乏有效治疗手段、致死率极高。Paxalisib是一种能穿过血脑屏障的PI3K/AKT/mTOR通路小分子抑制剂,于2018年进入II期临床试验。在2018年2月,FDA还授予了paxalisib治疗胶质母细胞瘤的孤儿药资格(ODD)。

今年4月初,Kazia公司公布了paxalisib治疗多形性胶质母细胞瘤(GBM)II期研究(NCT03522298)的阳性中期数据。该研究在新诊断的、非甲基化MGMT启动子状态的GBM患者中开展,正在评估患者接受最大限度手术切除和替莫唑胺(temozolomide,TMZ)联合同步放化疗后,将paxalisib作为辅助治疗药物的安全性、耐受性、推荐的II期剂量(RP2D)、药代动力学(PK)和临床活性。
研究结果显示,与现有标准护理TMZ相关的12.7个月相比,paxalisib辅助治疗的中位总生存期(OS)为17.7个月,具有明显的延长。paxalisib辅助治疗的中位无进展生存期(PFS)为8.5个月,长于TMZ相关的5.3个月。此外,接受治疗时间最长的患者,在确诊后19个月仍保持疾病无进展。目前,约半数入组患者仍在接受paxalisib治疗,随着研究的继续推进,OS和PFS数据可能进一步改善。
近二十年来,新诊断的胶质母细胞瘤患者没有任何新的药物治疗。以上临床数据表明,paxalisib正在迅速成为这一极具挑战性疾病全球管线中最有希望的候选药物之一。
Paxalisib(GDC-0084)合成工艺探索
Paxalisib(GDC-0084)实验室发现阶段的合成路线如图一所示。该路线以2,6-二氯-9H-嘌呤1为起始原料,通过选择性THP保护得到中间体2,然后用吗啉进行区域选择性取代,得到嘌呤3。随后使用强碱正丁基锂脱质子并与丙酮进行反应得到叔醇中间体4。对甲苯磺酸脱THP得到氨基醇5。而后,中间体5与2-溴乙酸乙酯进行亲核反应,随后酯水解提供二醇中间体6。二醇中间体6经环化得到环醚中间体7。最后,在经微波条件下的Suzuki交叉偶联反应得到目标产物GDC-0084。
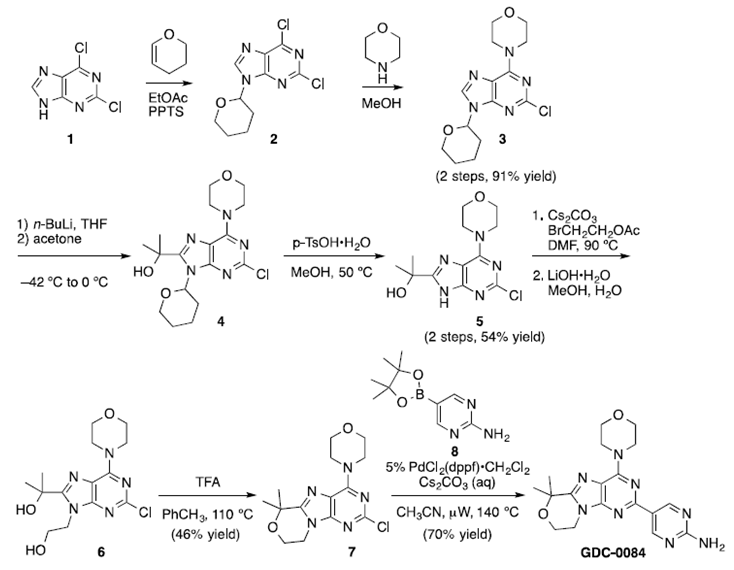
图一 实验室发现阶段合成路线
研究者分析该路线认为:①如果可以实现氨基醇5与合适的亲电试剂直接环合以制备吗啉中间体7,能够缩短2个步骤。并且也有可能降低中间体6到7的 SN1环化过程中因消除反应形成的杂质9。②路线中过度依赖柱层析色谱纯化步骤必须得到优化。③以Suzuki反应作为API合成的最后一步,需要解决API中残留重金属的问题。④API结晶鲁棒性需要进一步优化。
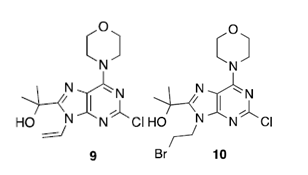
图二 路线中涉及的杂质
第一代工艺优化
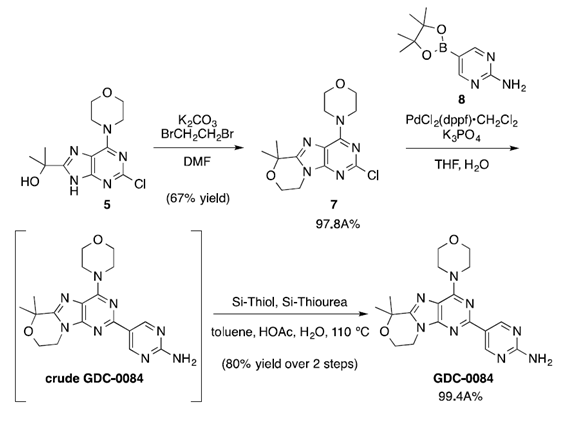
图三 第一代合成工艺路线
工艺优化的第一个目标是从叔醇中间体5直接转化得到吗啉杂环中间体7。经详细筛选,确定1,2-二溴乙烷为该环化反应的最优反应试剂。并进一步确定碳酸钾作为碱在DMF溶剂中进行该反应。由于该反应为放热过程,因此控制物料的加入速度是必要的。通过向1,2-二溴乙烷和碳酸钾的混合物中分批加入底物5,能够有效抑制分子间二聚体杂质的形成,且对转化率没有负面影响。
在确定了该步反应条件的基础上,研究者对中间体7分离纯化过程进行探索。向非均相反应混合物中加水未能诱导结晶,而是使产物呈油状析出。将产物萃取到乙酸乙酯中,并用水洗涤以除去DMF。然后将乙酸乙酯萃取液溶剂交换成2-丙醇,随后通过蒸馏结晶得到产物7。
最终,将该优化条件应用于5.83 Kg叔醇化合物5的环化反应,以67%的收率得到4.27Kg吗啉杂环7,HPLC纯度为97.8A%(杂质9为0.40A%)。
该合成步骤中存在四种潜在的遗传**杂质(溴乙烯,1,2-二溴乙烷,9和10)。通过开发气相色谱法对其进行检测,在最终分离得到的API中,这些杂质的定量均<6 ppm。
接下来,研究者对中间体7和芳基硼酸酯8之间的Suzuki交叉偶联反应进行详细研究,优化了反应的催化剂用量与反应溶剂。反应结束后,将反应液冷却至<10ºC使GDC-0084从反应混合物中析出,过滤得到滤饼并用水洗涤得到粗产品。利用优化后的反应条件,使用4.10 kg的中间体7成功进行了Suzuki交叉偶联反应,得到的产品能够在后续结晶步骤中进一步纯化。
粗品GDC-0084包含大量残留的钯,因此需要加入钯清除剂降低金属钯含量。仔细筛选后发现,在克级实验中,Si-Thiol和Si-Thiourea均可将残留的Pd降低至≤16ppm,而在公斤级实验中,则使用两种清除剂的混合物以确保有效去除不同状态的Pd。
在90ºC下,过滤除去清除剂并从2-丙醇中蒸馏结晶,最终以80%的收率和99.4A%的HPLC纯度提供了3.87kg的GDC-0084。
应用第一代合成工艺路线,研究者成功完成了3.87 kg GDC-0084的合成,用以启动临床研究。但是,该路线有几个方面需要进一步提高:①减少步骤1中消除副产物9的水平;②为步骤2确定更高效的催化剂;③最小化步骤3中乙酰胺杂质的形成。
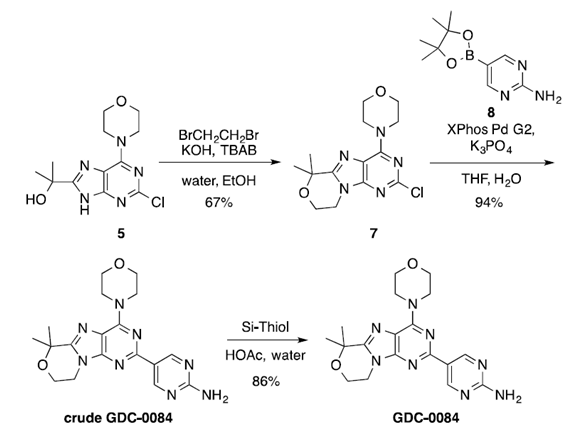
图四 第二代合成工艺路线
第二代工艺优化
在原工艺路线中,第一步关环反应会产生杂质9,并且由于使用DMF做溶剂,后处理过程需要溶剂交换,占用多个反应釜进行后处理,步骤繁杂。由此,研究人员开发了一种在水中使用相转移催化剂(PTC)的新工艺,用于合成中间体7。反应采用催化量的四丁基溴化铵(TBAB,30 mol%)在90ºC下反应 17 小时。反应结束后,产物呈油状从体系中析出,其中含有大量的1,2-二溴乙烷。通过加入乙醇,能够使产品从反应混合物中直接结晶析出,乙醇与水的最佳配比为1.3:1。利用该优化后的反应条件,在9.40 kg的规模下得到了6.85 kg的中间体7,收率为67%,高效液相色谱纯度为98.5A%,证明了该工艺的稳健性。
对于第二步的Suzuki偶联反应,主要问题在于钯催化剂用量过高以及总溶剂量大(58 vol)。经过仔细筛选,选择XPhos Pd G2作为催化剂,能够使催化剂的用量从2降至0.5 mol%。使用优化后的反应条件,以在6.75 kg 的中间体7作为底物,能够得到7.49 kg的粗GDC-0084(94%收率,99.4A% HPLC)。
API的纯化过程,通过进一步微调乙酸与水的比例,可以发现GDC-0084在90ºC时完全溶于10体积的乙酸:水(3:1)中,并在60ºC时结晶析出。研究者认为,30ºC温度差足以完成在90ºC进行的精滤过程(polish filtration)。 因为第二步中Pd的负载量从2mol%降低到0.5mol%,所以在该纯化步骤中用10wt%的Si-Thiol进行处理就足以将残留的Pd降低到10ppm以下。最终,在7.70 kg规模上验证了该纯化工艺,得到6.41 kg GDC-0084,HPLC纯度99.70A%,产率为83%。
第二代合成工艺通过3个步骤以52%的产率和99.70A%的纯度生产了6.41 kg的GDC-0084。基于PMI(process mass intensity)分析,与第一代合成方法相比,第二代合成方法有了显著的改进,并已被证明可以生产出公斤级高纯度原料药。
参考文献
1. Org. Process Res. Dev. 2016, 20, 4, 751-759.
2. ACS Med. Chem. Lett. 2016, 7, 4, 351-356.



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























