FDA过去专注于创新药物、器械和生物制剂发展所面临的挑战,然而仿制药开发同样面临各种科学挑战。本文将结合不同类型仿制药的开发,提供其关键路径。
PART
0 1.
仿制药开发流程
新药在研发及其临床试验的投入费用高昂,这些实验主要为了证明新药的安全、有效;为了鼓励新药研发,新药上市后将有一定期限的市场独占期。当新药的市场独占期或相关专利即将失效时,潜在的仿制药公司将对其开始进行仿制研究。仿制药获批的前提是证明其与参比制剂具有药学等效和生物等效。药学等效包括相同的API、相同剂量、相同剂型等。
生物等效是指仿制药活性成分在作用部位的吸收速度和吸收程度与参比制剂等同。大部分仿制药的药物等效和生物等效的研发路径简单明确,譬如分析化学可以鉴别和定量活性成分;通过比较药动学来证明生物是否等效。基于血液中药物浓度的生物等效评估是最常用的方法,成功应用于证明仿制药与参比制剂具有共同的安全性和有效性。
上述评估药学等效和生物等效的方法对复杂仿制药和局部用仿制药并不适用;科学上的挑战已经阻碍了该类仿制药的发展。
PART
0 2.
关键路径(Critical Path)
为了更有效的开发高质量的仿制药来提升公众健康,FDA确定如下四类关键路径:

上述方面的改善将加速仿制药的上市。更重要意义在于这将扩大仿制药范围的同时,确保仿制药安全、有效和质量可控。基于健全的科学评估等效性的方法,将建立医生、患者和公众对仿制药的信心。
1.质量源于设计(Quality by Design,QbD)
质量源于设计,通过理解和调整制剂处方和制造参数,将产品质量融入最终药品;最终检测仅用于确认药品的质量。仿制药公司可以通过改善制造流程让消费者获得高品质的仿制药。FDA鼓励将QbD应用于所有药品开发中,仿制药实施QbD将会遇到不同的挑战。
使用QbD来研发与参比制剂具有生物等效的仿制药,仿制药申请人必须理解含有特定活性成分药品的处方工艺参数可能对生物利用度的影响。
1.1 开发体内-体外(Vitro-In Vivo)相关性模型
目前制剂开发策略主要基于试错、内部数据库、研究人员的经验。通过建模和仿真可以实现制剂开发的系统化和机制化。药物吸收模型可用来评估(或预测)体内溶出或释放速率与药动学参数(用于评估生物等效性)之间的关系,这是评估不同处方工艺进而选择出最 佳制剂的有力工具。关键路径包括:
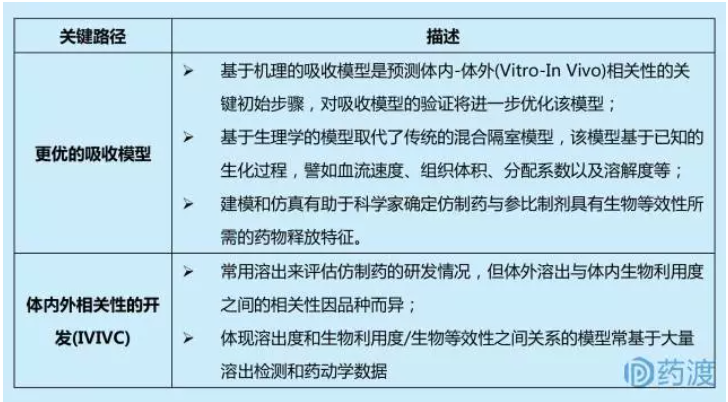
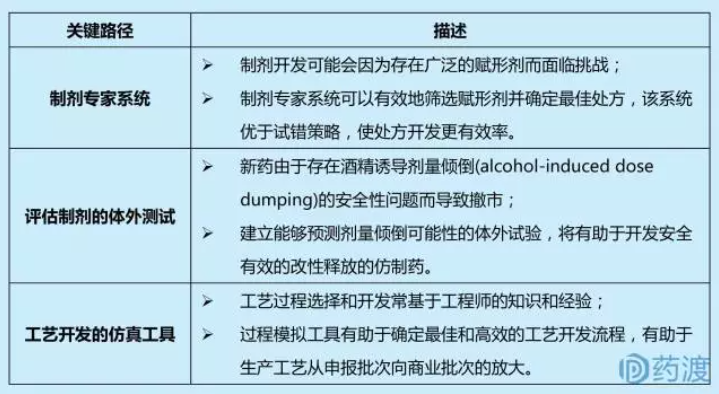
1.2 仿制药的处方和工艺开发
仿制药与参比制剂可以有不同的释放机理,前提是该仿制药和参比制剂具有药学等效和生物等效。仿制药可以利用QbD来确保新的释放机理和参比制剂具有生物等效。
ANDA申请人常用商业批的1/10做生物等效性研究。而ANDA获批后,申请人才会将批量放大至商业批。基于QbD理念,ANDA申请人通过识别出制造过程中的关键工艺参数,进而降低放大过程中失败的风险。关键路径包括:
2.全身作用仿制药生物等效性的方法
对于全身作用药物,关键路径目标是通过扩大生物等效性豁免,优化溶出方法和生物等效性的方法,来加速批准安全有效的仿制药。
2.1 扩大基于生物药剂学分类系统的生物等效性豁免
生物药剂学分类系统(BCS)是一种药物开发工具。对同时具备BCS I类(高溶高渗)和快速溶出的速释仿制药,申请人可提请生物等效性豁免。BCS II类(低溶高渗)和BCS III类(高溶低渗)同样可以申请生物等效性豁免。
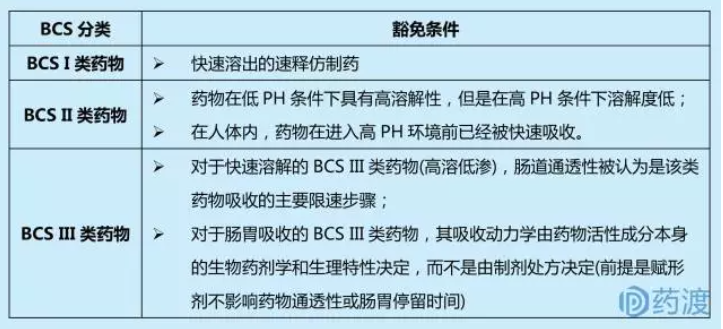
具有生物相关的溶出方法:开发生物相关的溶出方法,将为制剂研究人员提供与体内释放相关的可靠标准,同时允许监管者通过不同产品比较有意义的溶出曲线。
2.2 餐后生物等效性研究
即使参比制剂标签明确说明食物不影响药物吸收,当前FDA生物等效性指南仍然建议仿制药进行餐前餐后生物等效性的研究。餐后研究的目的是确保仿制药同样不存在食物影响,但对于快速溶出的BCS I类药物可豁免餐后生物等效性的研究。其关键路径包括:
食物和药物相互作用:若能够很好的理解药物和食物之间相互作用的机制,另外三种BCS类型药物亦可不需要进行餐后实验,以证明仿制药和参比制剂在餐后具有生物等效性。
2.3 新型给药技术的生物等效性
有别于传统速释口服片剂和胶囊剂,研究者持续开发了新型给药技术的仿制药。对于新型给药技术,需要进行额外的研究来改善生物等效性的评估。关键路径包括:
药代动力曲线含有多个峰的生物等效性:单次剂量的新型改良释放制剂(如皮下植入制剂)可在药时曲线中产生多个峰。FDA目前采用Cmax和AUC来评估生物等效性。开发评估生物等效性的新方法具有应用价值。
透皮产品:开发新的临床试验设计和标准,考察仿制药在皮肤刺激,潜在致敏性和附着性能与参比制剂的一致性。
2.4 高变异性仿制药的生物等效性
对于Cmax和AUC表现出个体高度变异性的药物,进行BE研究存在系列挑战。例如一种具有50%变异性的药物,即使仿制药和参比制剂是完全相同时,仍需要100例受试者才能证明其生物等效。除非高变异性药物具有宽治疗指数,否则这类药物既不安全也不有效。根据目前FDA指南,具有宽治疗指数的药物比窄治疗指数的药物更有研究价值。关键路径包括:
生物等效性研究设计:开发出仅需要更少受试者的生物等效性临床试验。
(未完,待续。下一章将重点介绍局部用药和靶向药物生物等效性的方法,包括吸入制剂、组合产品、皮肤病仿制药、脂质体仿制药等的生物等效性)。
参考文献:
1.FDA,Challenge and Opportunity on the Critical Path to New Medicinal Products (March 2004).
2.FDA,Critical Path Opportunities Report (March 2006).
3.FDA,Critical Path Opportunities List (March 2006).
4.Center for Drug Evaluation and Research,Approved Drug Products with Therapeutic Equivalence Evaluations (Orange Book),26th ed.(2006).
5.J. Woodcock,Biomarkers: Physiological & laboratory markers of drug effect,FDA (2006).
6.J. Woodcock,The concept of pharmaceutical quality,American Pharmaceutical Review p. 106 (November/December 2004).
7.Guidance for Industry:Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Basedon a Biopharmaceutics Classification System (August 2000).
8.Draft Guidance for Industry Bioavailability and Bioequivalence Studies for Nasal Aerosols and Nasal Sprays for Local Action (April 2003).



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57

























