与欧盟相比,美国生物类似药的发展起步较晚,但2018年绝对是美国生物类似药的丰收年,从2015年3月诺华山德士获批的第一个生物类似药Zarxio(filgrastim -sndz)至今,目前FDA共计批准了15种生物类似药,其中,在2018年获批的就达7种,创造了历史新纪录。此外,目前还有60多个生物类似项目正在进行中。
2018年7月18日FDA发布《生物类似药行动计划》(Biosimilars Action Plan),提出为鼓励生物制品创新和竞争以及生物类似药研发的系列举措;2018年12月11日,FDA局长Scott Gottlieb博士在一项声明也明确表示"美国目前正处于生物制品竞争市场发展的关键阶段,FDA将致力于推动科学和政策发展,以提高生物类似药开发效率"。
美国生物类似药法规是以2009年的《生物制品价格竞争和创新法》(BPCI)为基础,目前FDA关于生物类似药的法规指南主要包括:
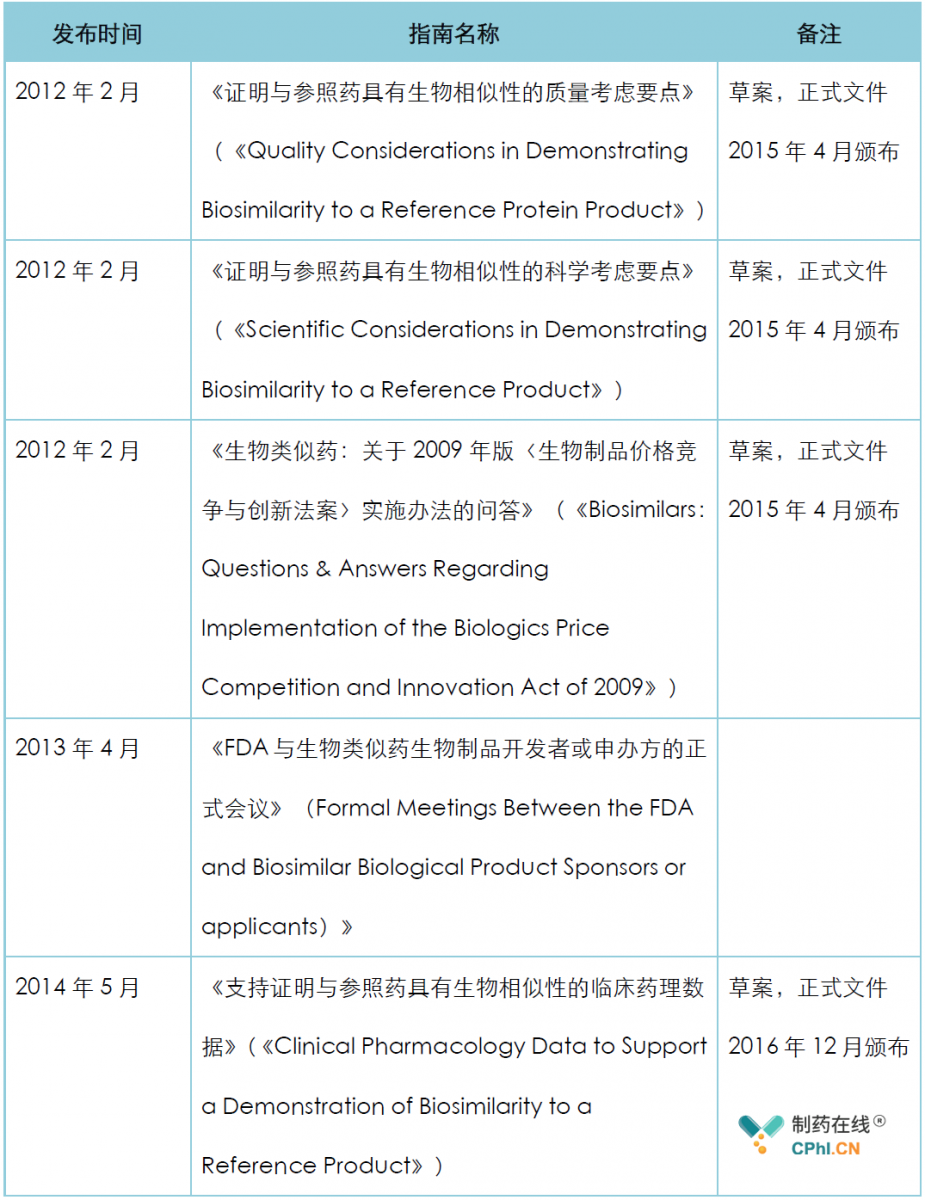
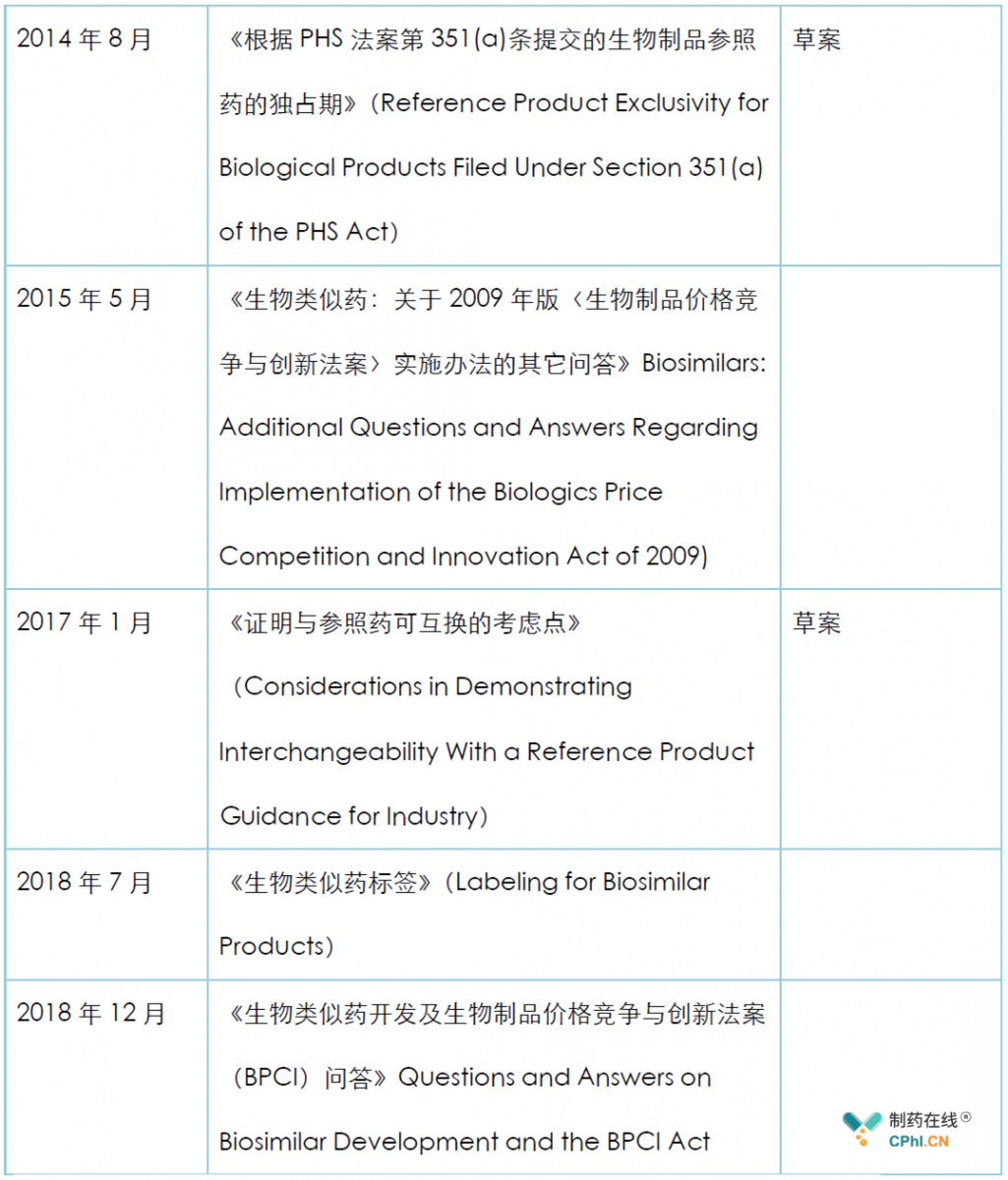
2018年12月,FDA发布了最新的生物类似药开发及生物制品价格竞争与创新法案(BPCI)问答,该问答指南修订了2015年4月颁布的《生物类似药:关于2009年版〈生物制品价格竞争与创新法案〉实施办法的问答》以及之后的其它问答,笔者对其中的内容作了翻译和整理:
★问题:I.1。主办方应该与谁联系,询问关于其生物类似药或可互换产品的开发方案的问题?
答:I.1。FDA在其网站上提供了当前的联系信息,参见FDA网站"Biosimilars," 从以下网站可获得https://www.fda.gov/biosimilars,并单击左栏中列出的链接"行业信息和指南"。
★问题:I.2。主办方应于何时要求与FDA召开会议,讨论其拟申报生物类似药或可互换产品的开发计划,以及主办方应向FDA提供哪些数据和信息作为本次会议的背景信息?
答:I.2。参见FDA指南草案:FDA与生物类似用户收费法案(BsUFA)产品主办方或申请者之间的正式会议,该指南草案描述了旨在促进生物类似开发项目的不同会议类型,以符合根据2017年生物相似用户费用修正案(BsUFA II)重新授权的2012年生物类似用户收费法案(BsUFA),以及支持该请求所需的标准/数据。要求的会议类型取决于产品开发阶段,以及在会议包中提交的信息是否符合会议类型的标准。
★问题:I.3。拟申报生物类似药可以与参照药的配方有差异吗?
答:I.3。拟申报产品与参照药之间配方的差异是可以接受的。一份351(k)申请必须包含证明生物产品与参照药高度相似的信息,尽管临床非活性成分有细微差异。此外,申请人需要证明生物产品和参照药在安全性、纯度和效力方面没有临床意义上的差异。例如,对于配方不含人血清白蛋白的拟申报产品,可以证实其与含人血清白蛋白的参照药生物相似。关于FDA对生物相似法定标准解释当前想法的更多信息,请参阅FDA的行业指南草案:证实治疗性蛋白产品与参照药生物相似的质量考虑要点以及证实与参照药生物类似的科学考虑要点。
★问题:I.4。拟申报生物类似药是否可以与参照药的给药装置或容器密封系统不同?
答:I.4。拟申报产品在给药装置或容器密闭系统设计上的一些差异可能是可以接受的。以下情况是可能的,例如,申请人拟申报生物类似药获批的是预灌封式或自动注射装置(这被认为是相同的剂型),即使参照药批准的是西林瓶包装,只要该产品符合生物类似性的法定标准并可提供给药装置或容器密闭系统足够的性能数据即可。拟申报产品在不同的给药装置或容器密闭系统,必须通过适当的研究,包括可提取/可浸出的研究和稳定性研究,证明其与最终的生物产品配方兼容。此外,对于给药装置或容器密闭系统的设计差异,可能需要性能测试和人为因素研究。
然而,给药装置或容器密闭系统的设计差异导致下列任何一种情况时,预期的生物类似药申请者将无法根据第351(k)条获得其产品的许可:
o 拟申报产品和参照药在安全性、纯度和效力方面有临床意义的差异;
o 不同的给药方式或剂型;或
o 使用参照药之前未获批准的条件(如适应症、给药方案);或者不符合生物相似性标准。
给药装置中的拟申报生物类似药将被视为组合产品,在某些情况下,该装置可能需要单独的申请。
拟申报可互换产品给药装置或容器密闭系统的信息,参见FDA指南草案:证实与参照药可互换性的考虑点。
★问题:I.5。申请人获得拟申报生物类似药批准时可否比批准的注射用参照药的给药途径少?
答:I.5。是的,申请人获得拟申报生物类似药批准时可以比批准的注射用参照药的给药途径少。申请人必须证实拟申报生物类似药与参照药在安全性、纯度和效力方面没有任何临床意义上的差异。在有限的情况下,可能需要提供采用未申请批准的给药途径的一项或多项研究的信息 (例如,采用皮下注射给药的一项研究可能会提供参照药和拟申报生物类似药更灵敏的免疫原性的比较评估,,即使拟申报生物类似药许可只申请了静脉注射给药途径)。
★问题:I.6。申请人获得拟申报生物类似药批准时可否比批准的参照药的所有表征要少(例如,规格、给药装置或容器密闭系统)?
答:I.6。申请人不需要获得批准的参照药所有表征的批准。然而,如果申请人想申请参照药特定适应症或与参照药对应的某一特定表征的适应症或使用条件的其它使用条件的批准,申请人可能需要该特定表征的批准(见问答I.4和I.5)。
★问题:I.7。申请人获得拟申报生物类似药批准时可否比批准的参照药的所有使用条件要少?
答:I.7。拟申报生物类似药申请人通常可以获得比参照药批准的所有使用条件更少的许可。351(k)申请必须包括以下证明信息:拟申报生物类似药提交的标签中规定、推荐或建议的使用条件或条件是参照药以前已被批准的(见PHS法案第351(k)(2)(A)(III)条)。
★问题:I.8。主办方是否可以使用非美国批准的产品所作的比较动物或临床数据,用于支持拟申报产品与参照药生物相似的论证?
答:I.8。主办方可以在某些研究中使用非美国批准的产品,以支持拟申报产品与美国批准的参照药生物相似的论证。然而,作为一项科学研究,用于支持生物相似性论证的分析研究和至少一项临床药动学(PK)研究,如果适当的话,至少一项药效学(PD)研究必须包括对拟申报生物类似药与美国批准的参照药直接进行充分的比较,除非有科学依据证明不需要这样的研究。
如果主办方按照PHS法案351(k)(2)(A)的规定想使用来自动物研究或比较拟申报产品与非美国批准的产品的临床研究的数据,主办方应提供足够的数据或信息,以科学地证明这些比较数据与生物相似性评估的相关性,并建立与美国批准的参照药之间可接受的桥梁。作为一种科学问题,所需的桥接数据类型将始终包括来自分析研究的数据(例如,结构和功能数据),这些数据直接比较所有三种产品(即拟申报产品,美国批准的参照药以及非美国批准的对照产品),可能还包括连接所有三种产品的临床PK和/或PD研究数据。这三种两两比较都应该满足分析和PK和/或PD相似度的预先指定的接受标准。这种办法的可接受性将根据具体情况加以评估,并应事先与FDA讨论。对于某些复杂的生物制品,可能需要改进方法。在审查申请的过程中,将对科学论证的充分性和桥接性作最后的确定。
在生物类似开发方案中,主办方在使用非美国批准的对照产品时可能需要解决的问题包括但不限于:
o 用于支持证实与美国批准的参照药使用条件和申请批准的患者人群相似性的临床项目设计的相关性;
o 非美国批准的对照产品许可持有人与美国批准的参照产品BLA持有人之间的关系;
o 非美国批准的对照产品是否在获得批准的设施中生产,并由具有类似FDA(例如,ICH国家)的科学和监管标准的监管机构进行检查;
o 非美国批准的对照产品是否由具有与FDA(例如,ICH国家)类似的科学和监管标准的监管机构批准,并且产品期限和范围是否已上市;和
o 非美国批准的对照产品与美国批准的参照药之间的科学桥梁包括理化特性、生物分析/功能分析、压力条件下的降解情况比较以及临床PK比较,并在适当情况下提供PD数据,以解决配方或初级包装的任何差异对产品性能的影响。
主办方还应考虑可能影响非美国批准的对照品与美国批准的参照药相似性评估比较数据的相关性的相它因素。
主办方可以提交有关非美国批准的对照产品的公开信息,以证明建立与美国批准的参照药之间桥梁所需要的比较数据的范围。产品的复杂性,特别是在高阶结构方面,翻译后修饰(如糖基化)以及与产品相关的异质性程度,可能会影响对桥接数据范围的科学论证的考虑。FDA可能考虑的关于桥接数据范围的其他因素包括,但不限于:
o 美国批准的参照药和非美国批准的对照产品的的配方、剂型和规格是否相同;
o 美国批准的参照药和非美国批准的对照产品的给药途径;
o 物理、化学和生物/功能评估的设计以及使用足够灵敏的多重正交方法检测产品之间的差异;
o 选择用于建立科学桥梁的非美国批准的对照产品批次的科学依据,以及如何选择非临床和临床研究中使用材料的批次。这座科学桥梁应该包括足够批次的非美国批准的对照产品,以充分捕获产品质量属性的可变性。如果可能的话,在非临床或临床研究中使用的非美国批准的对照产品的批次应包括在建立分析桥梁的评估中。
鼓励主办方在开发项目期间与FDA讨论科学论证的充分性以及与美国批准的参照药之间的联系桥梁。FDA将在对351(k)申请进行审查期间,对这一科学论证和桥梁的充分性做出最终决定。
有关非美国批准的对照品是否可用于旨在支持确定与参照药可互换性所需的附加标准的研究中的更多信息,请参见FDA的工业指南草案"证实与参照药可互换性的注意事项"。
作者简介:zhulikou431,高级工程师、PDA会员、ISPE会员、ECA会员、PQRI会员、资深无菌GMP专家,在无菌工艺开发和验证、药品研发和注册、CTD文件撰写和审核、法规审计、国际认证、国际注册、质量体系建设与维护领域,以及无菌检验、环境监控等领域皆具有较深造诣。近几年开始着力关注制药宏观领域趋势分析和制药企业并购项目的风险管理工作。




合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57





















