FDA作为全球 领先的药品审评和监管机构,其工作计划对于医药行业从业者会产生明显影响,因此也为行业所关注。每年,FDA会发布新一年的技术指南修订和制订计划,来说明自己的工作重点。本文将结合2025年FDA CBER指南工作计划和CDER指南工作计划来介绍,希望可以为中国制药行业提供参考。
第一部分:CBER2025年指南工作计划整体情况
近期,FDA官网发布《Guidance Agenda: Guidance Documents CBER is Planning to Publish During Calendar Year 2025》,对FDA下属CDER将在2025年发布的指南,进行了介绍;详情参见下表。

第二部分:CBER2025年指南工作计划重点介绍
下面对这份2025年技术指南工作计划,选择重点内容进行介绍。
◆Blood and Blood Components
※Recommendations for the Evaluation of Blood Collection, Processing, and Storage Devices Using Non-Di(2-ethylhexyl) Phthalate (non-DEHP) Materials
翻译:使用非邻苯二甲酸二(2-乙基己基)酯(非DEHP)材料的血液采集、处理和储存器械评估建议。
血液 制品是高风险产品,其所使用的包材或者容器材质对于质量影响很关键。因此,对于企业采用新材质,都需要进行充分评估。这份指南将提供评估思路。
◆Therapeutic Products
※Frequently Asked Questions — Cell and Gene Therapy Products
翻译:细胞和基因治疗产品常见问题
细胞和基因治疗产品是这几年国际和美国国内研发热点,有很多问题需要厘清。FDA发布这份指南,将促进相关问题的认知深化。这份指南编制工作在2024年已经被列为工作计划之一,没有完成,这次仍然被列为2025年工作计划之中。
※Post Approval Methods to Capture Safety and Efficacy Data for Cell and Gene Therapy Products
翻译:用于获取细胞和基因治疗产品的安全性和有效性数据的批准后方法
细胞和基因治疗产品属于新型药品,临床使用情况比较特殊。在这类药品获得批准上市后,MAH如何规范收集安全和有效数据,需要官方更明确要求。这份指南将解决这类问题。
第三部分:CDER2025年指南工作计划整体情况
近期,FDA官网发布《CDER Guidance Agenda
New and Revised Draft Guidances Planned for Publication in Calendar Year 2025》,对FDA下属CDER将在2025年发布的指南,进行了介绍;详情参见下表。
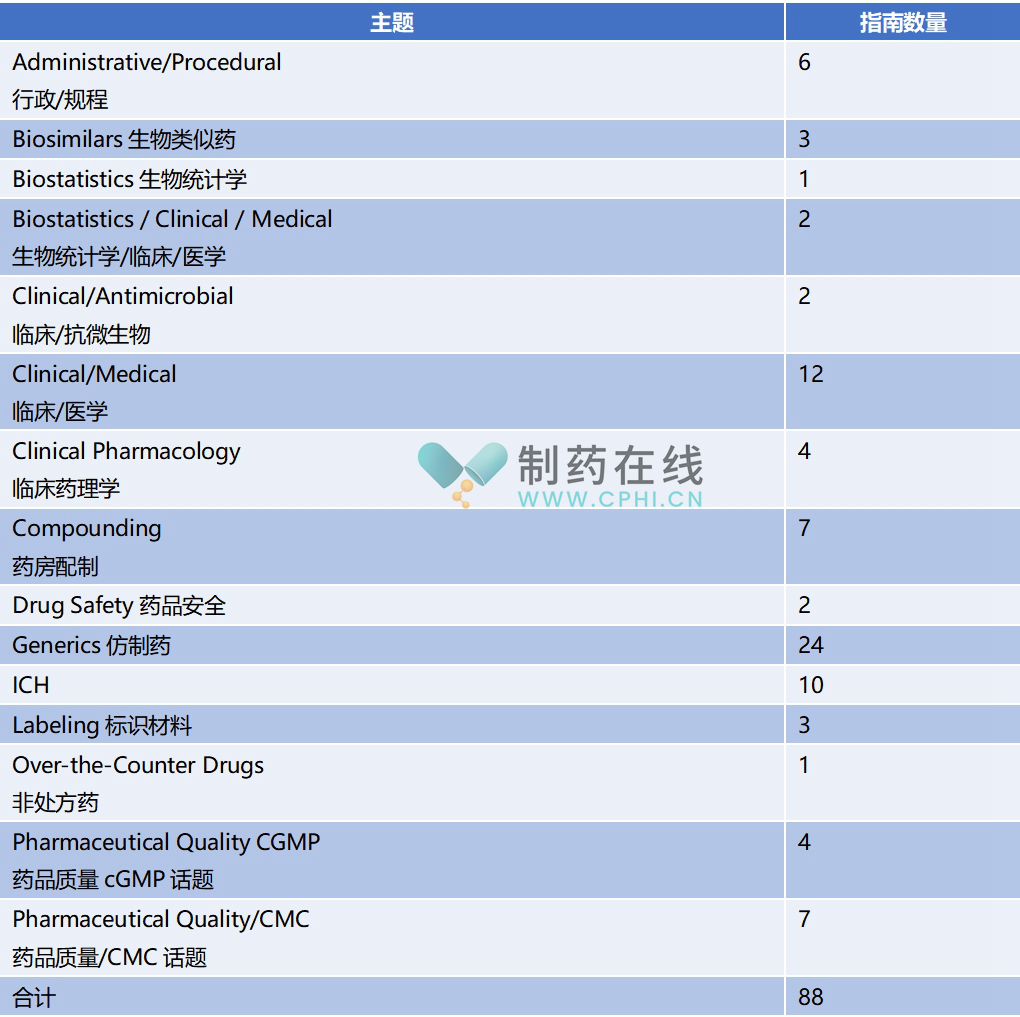
分析:和过去历年CDER工作计划安排类似,FDA CDER在2025年技术指南修订和制订工作计划中,重点工作领域还是临床医学、仿制药、ICH专题和CMC专题等。
第四部分:CDER2025年指南工作计划重点介绍
下面对这份2025年技术指南工作计划,选择重点内容进行介绍。
◆Administrative/Procedural专题
※Civil Monetary Penalties for Failure to Meet Accelerated Post Marketing Requirements
翻译:未能满足加速上市后要求的民事罚款
FDA为了促进创新药上市,设置了优先审评程序。但是这些通过优先审评程序上市的药品,需要按照官方要求完成上市后部分补充研究任务。而部分企业只想享受待遇,而不想尽到责任;就存在采用优先审评程序上市后不规范开展补充研究的情况。FDA将在2025年发布相关指南,督促企业完成。
※NDC Creation, Assignment, Listing and Appropriate Use for Human Drugs, Including Biological Products
翻译:人用药物(包括生物制品)的NDC创建、分配、登记和适当使用指南。
对于在美国市场流通的药品,都需要向FDA申请NDC号码;这是用于产品生命周期追溯的一个关键号码。但是提醒读者注意,这个号码是用于追溯的,不代表产品获得FDA审核或者批准。这个号码和国内批准文号不是一个概念。
◆Biosimilars专题
※Biosimilar and Interchangeable Biosimilar Products: Considerations for Container Closure Systems and Device Constituent Parts
翻译:生物类似药和可互换生物类似药产品:容器密封系统和器械组成部分的注意事项。
生物类似药和参比生物制品的可比性关系,远超过化学仿制药和化药参比制剂的关系。
为了证明生物类似药满足技术要求,可以在临床上替代参比生物制品,每个国家审评机构都设置了很多技术要求。
生物类似药要上市,需要和参比生物制品进行头对头对比。FDA发布这份新指南,将指引企业在研发生物类似药方面,关注包材的可比性。
◆Clinical/Medical专题
※Considerations for the Inclusion of Older Adults in Clinical Trials
翻译:将老年人纳入临床试验的考虑因素
FDA发布这份指南,将指导企业在受试者范围涵盖老年人时,需要考虑哪些影响因素;将促进临床试验的规范开展。
◆Generics专题
※180-Day Exclusivity: Questions and Answers
对于首仿药给与180天的行政保护,这项措施已经实施多年。为了更细致解释这项政策,FDA将出台配套指南。这项工作被列入2024年指南工作计划,未完成。FDA将在2025年工作计划中,继续推荐这份指南。
※Refuse-to-Receive
ANDA Submissions-Refuse-to-Receive for DMF Facilities Deficiencies
翻译:DMF中设施缺陷导致的ANDA申报的拒绝接受
ANDA Submissions-Refuse-to-Receive Standards: Questions and Answers
翻译:ANDA申报拒绝接受标准问答。
FDA为了提高ANDA申报效率和质量,发布了很多份RTR指南;这些指南为行业指明了方向,需要企业技术人员反复研读。
※ANDAs for Certain Highly Purified Synthetic Peptide Drug Products That Refer to Listed Drugs of rDNA Origin
翻译:仿制rDNA来源参比制剂的某些高纯度合成肽药品的ANDA。
中国一致性评价工作正如火如荼的开展,目前一致性评价范围覆盖口服药品和注射剂2个大类。根据2025年初国务院的53号文,2025年中国将把一致性评价药品范围拓展到贴剂等范围。
而美国仿制药的范围已经拓展到rDNA来源参比制剂为被仿制对象的合成多肽类仿制药;因此不得不佩服FDA对药品研发和监管科学的推动工作。
※半固体制剂的技术指南
In Vitro Permeation Tests for Semisolid Topical Products Submitted in ANDAs
翻译:在ANDA中提交的半固体局部产品的体外渗透测试指南。
In Vitro Release Tests for Semisolid Topical Products Submitted in ANDAs
翻译:在ANDA中提交的半固体外用产品的体外放行测试指南。
半固体制剂属于特殊剂型,对于这类产品的研发和注册,FDA已经发布了很多PSG来指导。在2025年,FDA将发布2份指南,阐释类似问题。
◆ICH专题
※M4Q(R2) Addressing Common Technical Document (CTD) Quality-Related Questions
翻译:ICH M4Q(R2) 解决通用技术文档 (CTD) 质量相关部分的问题
ICH M4已经发布多年,为国际药品注册和贸易发挥了重大作用。然而,现行版CTD-Q部分确实太陈旧了,从格式看主要适用于化药和重组类生物制品,不适用于近几年研发热点,例如细胞基因治疗产品、药品-器械组合产品等。这次修订,将对类似问题进行阐释。
※Q1/Q5C Targeted Revisions of ICH Stability Guidelines
翻译:ICH Q1/Q5C-稳定性试验指南修订
这份指南很重要,是药品研发和注册阶段非常重要的内容。然而,由于过去国内对这些工作不重视,导致国内企业很多人员对于类似技术信息不关注。根据网络流传信息,ICH将把ICH Q1系列指南修订为核心指南+配套附录的组织形式。
◆Pharmaceutical Quality CGMP
※Responding to Form FDA 483 Observations at the Conclusion of a Drug CGMP
翻译:对药品CGMP结论的FDA 483表格观察项的回应指南
FDA在对各类企业检查后,如果有缺陷,会签发一份表格,就是行业内都关注的483表格。如果企业回复483所列缺陷不能令FDA满意,或者回复时间较晚,就会被FDA警告。过去FDA针对如何回复483无具体要求,因此行业内人士对此问题很困惑。这份指南编制工作在2024年已经被列为工作计划之一,没有完成,这次仍然被列为2025年工作计划之中。
◆Pharmaceutical Quality/CMC
※Container Closure Systems for Drugs, Including Biological Products
翻译:药品和生物制品容器密封系统指南
这份指南上一版本还是1999版,到现在已经26年了。虽然从内容看,这份1999年指南还算齐全,但是毕竟行业发生了巨变,这份老指南需要被修订。这份指南编制工作在2024年已经被列为工作计划之一,没有完成,这次仍然被列为2025年工作计划之中。




合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57





















