
这是一组回顾过去五年中国药政体系演变的文章,也算蹭流量的文章。如果你见识深刻,立意高远,请不要看,这组文章估计会浪费你的宝贵时间。
本文是这组系列文章的一部分,主要介绍中国大陆地区原辅包关联审评政策演变进化的历程;如果从2016年算起,到如今这段历程看,这篇文章题目应该是九年风雨回顾。为了和其他系列文章保持一致,因此题目没有调整。希望此文可以让制药同仁经常回望走过的路,以资借鉴。
说明:本文所涉及包材,指的是直接接触药品的内包材;为了行文方便,以后都称为内包材。
第一部分:原辅包关联审评之前的管理状态
A1-原料药管理状态
先看《药品管理法》对药品定义规定:
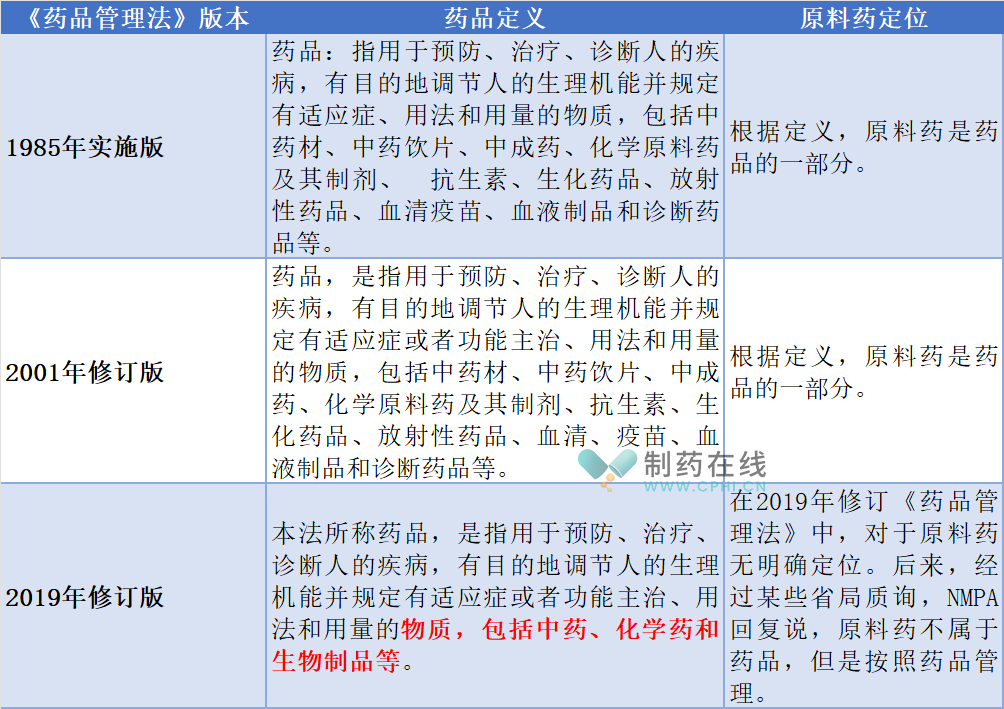
分析:综合上面信息,目前化学原料药在中国大陆地区不是药品,但是按照药品管理。
A2-辅料管理状态
关于辅料过去管理的法规条款,参见下表:

分析:在过去多个版本的《药品注册管理办法》中,都提到需要提供辅料合法来源的证明文件。
A3-内包材管理状态
关于药品包材过去管理的法规条款,参见下表:
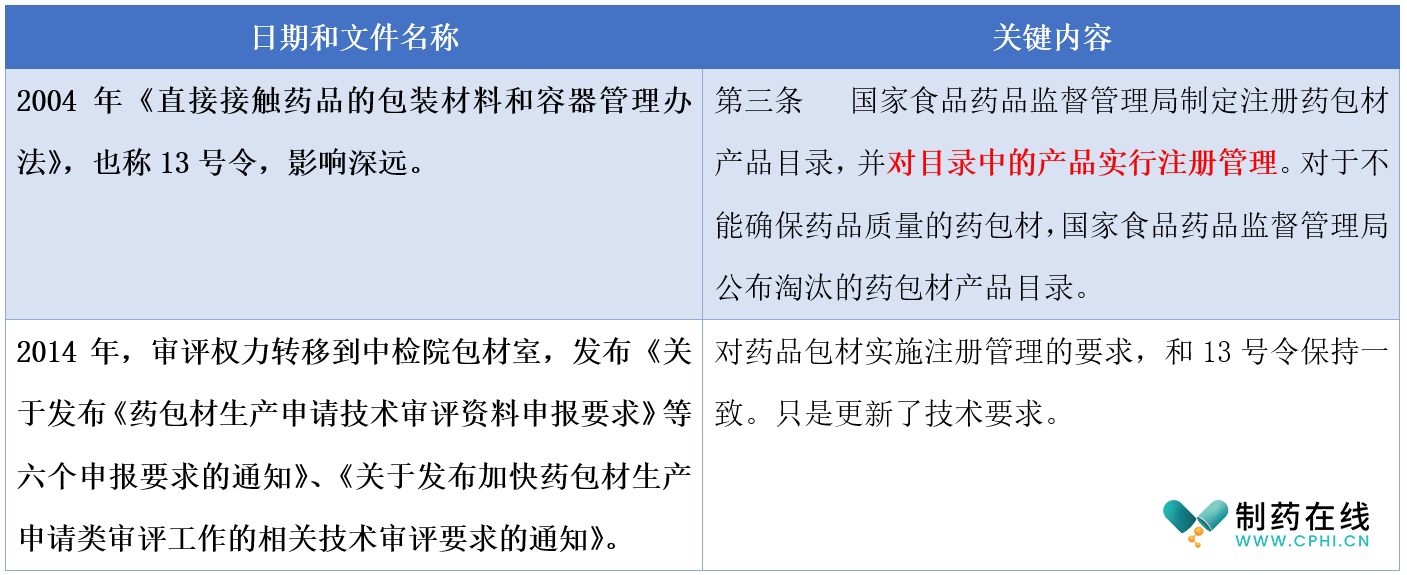
分析:在实施原辅包关联审评之前,包材也是实施注册制。
第二部分:2016年辅料包材关联审评肇始
20160810,发布《总局关于药包材药用辅料与药品关联审评审批有关事项的公告》(2016年第134号)。
三、自本公告发布之日起,药包材、药用辅料应按程序(见附件2)与药品注册申请关联申报和审评审批,《药包材及药用辅料申报资料要求》另行公布。各级食品药品监督管理部门不再单独受理药包材、药用辅料注册申请,不再单独核发相关注册批准证明文件。
分析:根据2016年134号公告,中国药政历史上的原辅包关联审评制度开始建立。
第三部分:2017年原辅包关联审评政策全面推进
20171130,总局发布《总局关于调整原料药、药用辅料和药包材审评审批事项的公告》 (2017年第146号) 。
为贯彻落实中共中央办公厅、国务院办公厅《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号)与《国务院关于取消一批行政许可事项的决定》(国发〔2017〕46号),取消药用辅料与直接接触药品的包装材料和容器(以下简称药包材)审批,原料药、药用辅料和药包材在审批药品制剂注册申请时一并审评审批。现就有关事项公告如下:
一、 药品注册申请人在中华人民共和国境内提出的注册分类2.2、2.3、2.4、3、4、5类药品制剂申请所使用的原料药,以及各类药品注册申请所使用的药用辅料、药包材适用于本公告要求。
分析:从这个文件开始,关联审评制度全面覆盖的原料药、辅料和包材的每个领域。
第四部分:2019年原辅包关联审评最全奠基
20190716,国家局发布《国家药监局关于进一步完善药品关联审评审批和监管工作有关事宜的公告》 (2019年 第56号)。
一、总体要求
(一)原辅包的使用必须符合药用要求,主要是指原辅包的质量、安全及功能应该满足药品制剂的需要。原辅包与药品制剂关联审评审批由原辅包登记人在登记平台上登记,药品制剂注册申请人提交注册申请时与平台登记资料进行关联;因特殊原因无法在平台登记的原辅包,也可在药品制剂注册申请时,由药品制剂注册申请人一并提供原辅包研究资料。
(三)药品制剂注册申请人申报药品注册申请时,需提供原辅包登记号和原辅包登记人的使用授权书。
(四)药品制剂注册申请人或药品上市许可持有人对药品质量承担主体责任,根据药品注册管理和上市后生产管理的有关要求,对原辅包供应商质量管理体系进行审计,保证符合药用要求。
分析:在2019年这个最新规定里面,对于原辅包关联审评政策,进行了重新梳理和规定。并且,对于已在中国上市药品中使用的原料药,允许单独登记获得A的登记状态。
第五部分:2023年原料药再注册的规定
20231013,发布《国家药监局关于化学原料药再注册管理等有关事项的公告(2023年第129号)》。
一、 总体要求
(一)化学原料药按照药品管理,其登记注册、补充申请及再注册申请属于行政许可事项,药品监管部门按照《药品管理法》《药品管理法实施条例》《药品注册管理办法》等有关规定开展审评审批。境内生产化学原料药登记人应为化学原料药实际生产企业,境外生产化学原料药登记人应委托中国境内的企业法人进行登记。
分析:根据上面规定,可以得到2个结论:
---化学原料药不是药品,但是按照药品管理。
---中国大陆地区,化学原料药不允许委托生产,生产企业就是登记人。
(六)为有序开展化学原料药再注册工作,给予一定过渡期。自本公告发布之日起,化学原料药批准证明文件剩余有效期在6个月以上的,按照上述第(五)项规定申请再注册;批准证明文件剩余有效期不满6个月或已过有效期的,登记人应在本公告发布之日起一年内,向省级药品监管部门(或药审中心)提出再注册申请。
分析:化学原料药登记人需要向所在地省局申请再注册。
第六部分:2025版药典的影响探讨
影响1-药典标准适用性问题
如果到2025年初,一个制药企业技术人员还认为,《中国药典》上的标准会适用于生产同类品种的所有企业,那就相当荒唐了。药典收载的质量标准仅仅适用于某些生产工艺生产的品种。如果生产工艺差距较大,质量标准就可能不会适用。
2024年1月1日生效的《药品标准管理办法》提到:
第二十五条新版国家药品标准颁布后,持有人经评估其执行的药品标准不适用新颁布的国家药品标准有关要求的,应当开展相关研究工作,按照药品上市后变更管理相关规定,向药品审评中心提出补充申请并提供充分的支持性证据。符合规定的,核准其药品注册标准。
第三十一条新版国家药品标准颁布后,执行药品注册标准的,持有人应当及时开展相关对比研究工作,评估药品注册标准的项目、方法、限度是否符合新颁布的国家药品标准有关要求。
对于需要变更药品注册标准的,持有人应当按照药品上市后变更管理相关规定提出补充申请、备案或者报告,并按要求执行。
分析:从上面这2条内容看,药典收载的质量标准适用性,是需要评估确定的。
影响2-辅料的元素杂质和残留溶媒控制要求
◆20250102,发布《国家药用辅料标准编写细则》 (2025年版)。关于辅料中元素杂质控制要求,规定如下:
通则0251中规定:药用辅料残留溶剂和元素杂质的控制应参照相关通则(通则0861和0862)和ICH的要求,根据药用辅料的生产工艺和拟应用的制剂需要,对药用辅料中的残留溶剂和元素杂质进行风险评估和相应控制,以确保药用辅料的质量、安全及功能满足制剂的需要。基于风险评估确定需要在标准中列入检查项的,应采用适宜的经过验证的分析方法检测。药用辅料元素杂质风险评估信息表可用于元素杂质风险评估与控制的确认。药品已经按照相关要求进行了元素杂质评估和控制的,其药用辅料可不需要再按《中国药典》药用辅料品种正文中的元素杂质相关检查项目(包括重金属、砷盐及其他ICH Q3D表5.1中的元素的相应检查项目)进行检测来证明符合规定。《中国药典》药用辅料品种正文中设置残留溶剂和元素杂质项目与否,药用辅料均应符合所关联制剂的要求。
◆20250102,发布《国家药用辅料标准编写细则(2025年版)》。下面是涉及辅料中残留溶媒控制要求。
《中国药典》药用辅料品种正文在确定是否收载残留溶剂检查项时,可参考如下原则:
①《中国药典》收载的药用辅料的残留溶剂如包含1类溶剂(本文中溶剂分类均为ICH Q3C最新版本的分类,以下同此原则),应在品种正文中设置残留溶剂检查项,并列明检测方法和限度。
如有企业该品种不涉及上述残留溶剂,在该项目后增加限制条件,即在相应项目后备注如“(生产工艺中使用或产生时测定)”。
②《中国药典》收载的药用辅料的残留溶剂如包含2类、3类,一般不在品种正文中设置残留溶剂检查项。
如产品中残留量较高,或风险较高,应在标示项中要求标示相应残留溶剂,即“【标示】应标示本品中残留溶剂***的限度”。
如上述残留溶剂检测中涉及特殊的前处理方法或测定条件,可在品种正文后的“附”下列出测定方法。
③《中国药典》收载的药用辅料的残留溶剂如为ICH Q3C中没有足够毒理学数据的溶剂或未列入的溶剂,如环氧乙烷等,应通过对溶剂的安全性、产品的使用情况、工艺控制水平以及国内外药典控制情况进行多方评估,必要时在品种正文中设置检测项目和限度。
第七部分:疑难问题讨论
问题1-一个登记号是否包含多种生产工艺?
关于这个问题,国际上管理要求发生过剧烈变化。
◆2018年7月亚硝胺杂质事件之前
在此之前,US DMF制度和欧盟ASMF/EDQM CEP申报资料,都允许:在确保关键中间体和API质量标准保持一致的情况下,允许一个申请人在一份DMF中提交多种生产工艺。
◆2018年7月亚硝胺杂质事件之后
在亚硝胺杂质事件以后,鉴于这个事件暴露出的弊端,各国都对API申报中包含多种生产工艺的情况进行了限制。
2024年3月,EDQM发布《Content of the dossier for CEP applications for chemical purity and microbiological quality of substances for pharmaceutical use》。其中提到:
Whatever type of manufacturing process is used, alternatives within the same dossier are only allowed if not substantially different. Even if the quality of late stage key intermediates and final substance from the alternative process are not affected in terms of specification and impurity content but the processes are substantially different, they cannot be accepted in the same application. A separate CEP application covering the same substance with the difference(s) explained in a subtitle may need to be submitted for each alternative process.
翻译:无论使用哪种类型的制造工艺,只有在没有实质性差异的情况下,才允许在同一申报资料中使用替代工艺。即使替代工艺的后期关键中间体和最终物质的质量在标准和杂质限度方面不受影响,但工艺存在实质性差异,也不能在同一申请中接受它们。对于每个替代工艺,可能需要提交涵盖相同物质的单独CEP申请,并在副标题中解释这种差异。
◆在国内,2023年7月3日,国家局发布《化学原料药受理审查指南(试行)》提到:

问题2-辅料和包材允许委托生产吗?
这个问题,曾经出现过曙光。20240718,NMPA发布《国家药监局综合司公开征求《关于发布〈药用辅料生产质量管理规范〉〈药包材生产质量管理规范〉的公告(征求意见稿)》意见》。其中提到:

但是,到了2025年1月2日,NMPA发布《国家药监局关于发布《药品生产质量管理规范(2010年修订)》药用辅料附录、药包材附录的公告》(2025年第1号)。在这份最新文件中,取消了类似描述。
问题3-辅料和包材如何规范管理变更?
2025年1月2日,NMPA发布《国家药监局关于发布《药品生产质量管理规范(2010年修订)》药用辅料附录、药包材附录的公告》(2025年第1号),提到:(二)严格变更管理。药用辅料、药包材生产企业应当按照药用辅料附录、药包材附录等要求,建立变更管理体系,根据风险确定药用辅料、药包材生产过程中变更的类别,开展相应研究,由质量管理部门批准后方可实施,并更新药审中心原辅包登记平台信息,及时告知药品上市许可持有人。
对于可能影响药用辅料、药包材质量的变更(如生产工艺、原材料来源、生产场地等变更),应当在研究过程中与药品上市许可持有人充分沟通。
但是时至今日,国家药监局未发布针对辅料企业自身和包材企业自身的变更指导原则。因此,如果要启动变更,辅料企业需要参考IPEC变更指南来评估管理;而药品包材企业要参考中国医药包装协会指南来实施变更管理。
系列文章推荐:
作者简介:zhulikou431,高级工程师、PDA会员、ISPE会员、ECA会员、PQRI会员、资深无菌GMP专家,在无菌工艺开发和验证、药品研发和注册、CTD文件撰写和审核、法规审计、国际认证、国际注册、质量体系建设与维护领域,以及无菌检验、环境监控等领域皆具有较深造诣。近几年开始着力关注制药宏观领域趋势分析和制药企业并购项目的风险管理工作。




合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57





















