首先说明,如果你对无菌制药法规很熟悉,对于无菌制药工艺很熟悉,请不要阅读此文,估计会浪费你宝贵时间。
近期,网络公开流传中国2010版GMP附录1无菌药品内部稿,参见下面截图:

为了让行业人士了解无菌GMP法规的最新进展,以及这部正征集意见的无菌GMP附录对中国无菌药品的影响,特撰写此文,分享自己的看法和认知。另外,为了行文方便,本文将以技术专题方式来展开。
专题讨论01-中国无菌GMP附录1内部稿适用范围
为了清晰对比,可以看下面表格:

分析:在GMP附录1适用范围方面,中国GMP附录1无菌药品征集意见稿基本上完全对标EU GMP附录1(2022版);但是要求内容位置进行了调整,调整到后面的244条。
提醒国内生产类似产品(例如注射级辅料、注射级原料药、液体产品)的企业,也需要学习和评估GMP附录1无菌药品的影响。
专题讨论02-CCS影响范围有哪些
经过梳理,在这份征集意见稿里面,如下条款提到污染控制策略(CCS)。
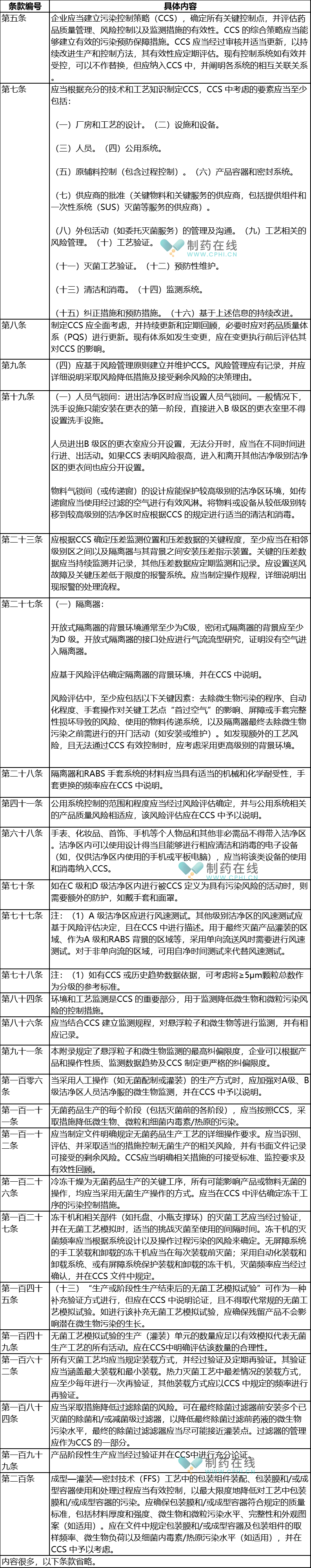
分析:从上面表格内容可以看出,附录1无菌药品征集意见稿引入CCS概念,而且覆盖了影响产品微生物、内毒素、颗粒相关的领域。
专题讨论03-隔离器分类和RABS分类
根据附录1无菌药品征集意见稿的第二百四十五条术语部分的定义,封闭式隔离器和开放式隔离器定义如下:
◆密闭式隔离器系统通过与辅助设备的无菌连接实现物料转移,而不是使用向周围环境开口的方式,从而阻断外部污染物进入隔离器内部。密闭系统在整个操作中保持密封状态。
◆开放式隔离系统允许在操作过程中通过一个或多个开口连续或半连续地传进和进出物料。开口的设计(如使用持续正压)以阻断外部污染进入隔离器。
这部附录1无菌药品征集意见稿没有对RABS进行分类,然而根据2025年1月FDA发布的《Outsourcing Facility Inspections》提到:
Operators use glove ports, half suits or automation to access areas within the enclosure during filling. There are two types of RABS, “open” and “closed” RABS. The doors to a “closed” RABS are never opened during an operation. While an “open” RABS is designed to operate with doors always closed, on rare pre-defined circumstances the doors of the enclosure can be opened to perform certain interventions.
翻译:在灌装过程中,操作员使用手套端口、半身衣服或自动化装置进入外壳内的区域。RABS有两种类型,“开放类型RABS”和“封闭类型RABS”。在操作过程中,“封闭类型RABS”的门永远不会打开。虽然“开放式RABS”设计为在门始终关闭的情况下运行,但在极少数预先定义的情况下,可以打开外壳的门以便于执行某些干预动作。
分析:无菌企业应该结合上述法规和指南,在建立CCS、采购关键设备和建立操作规程时,对自己使用的关键设施进行明确和准确定义、管理。
专题讨论04-洁净服管理
具体内容参见下面表格:

分析:从上面内容可以看出,如果要符合这些要求,制药企业洁净区人流通道需要重新布局;对洁净服供应商管理需要加强。
专题讨论05-SUS系统管理
这版附录1无菌药品征集意见稿增加了一个单独章节,专门介绍一次性系统的管理要求,内容从225条到233条,共9条内容。
对于一次性系统的验收,侧重于供应商评估和审计。另外,在对入厂的SUS系统验收时,需要分为入库第一次验收和车间使用时的第二次验收。
专题讨论06-BFS产品的管理
针对这种工艺,需要关注如下内容:
◆聚合物原料的质控要求
例如第二百一十二条应验证挤出系统为成型容器提供适当的无菌保证的能力。应在文件中定义和控制聚合物原料的取样频率、微生物负荷以及细菌内毒素/热原水平(如适用),并在CCS中予以考虑。
◆无菌工艺BFS的人员更衣要求
例如第二百零八条用于生产非最终灭菌产品的BFS 设备应符合以下要求:
(一)用于无菌灌装的往复式设备,型胚在环境中敞口,因此型坯挤出、吹塑和封口的关键区域、灌装环境的设计和维护应满足A 级条件。
(二)用于无菌灌装的旋转式设备,型坯通常在成型后密闭于环境,型坯内的灌装环境的设计和维护应满足A 级条件。
(三)设备应至少安装在C 级环境中,且操作人员应穿着A/B 级洁净服。
分析:按照上面最新要求,BFS生产线需要对这个更衣特殊要求,进行人流通道重新布局,设置穿戴A/B级洁净服的房间。
总结
综上所述,无菌企业需要高度关注这部正在征集意见的附录1文件,并从现在开始,在建设无菌厂房、采购关键设备、寻找无菌CDMO开始,全面提升技术要求,避免以后被动。
作者简介:zhulikou431,高级工程师、PDA会员、ISPE会员、ECA会员、PQRI会员、资深无菌GMP专家,在无菌工艺开发和验证、药品研发和注册、CTD文件撰写和审核、法规审计、国际认证、国际注册、质量体系建设与维护领域,以及无菌检验、环境监控等领域皆具有较深造诣。近几年开始着力关注制药宏观领域趋势分析和制药企业并购项目的风险管理工作。




合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57





















