2019年底修订生效的《药品管理法》对于药品上市后的进一步研究做出了明确规定:

同样,在2020年7月1日生效的《药品注册管理办法》也提到类似要求:

第六十六条 对附条件批准的药品,持有人应当在药品上市后采取相应的风险管理措施,并在规定期限内按照要求完成药物临床试验等相关研究,以补充申请方式申报。
从上面的法规内容可以看出,一个MAH在自己产品获得批准后,还不能躺在功劳簿上面休息,还需要承担生命周期内的各类合规任务。
根据CDE公开的数据,据不完全统计,2020年上半年NMPA(国家药品监督管理局)共批准 27 个新药上市,其中18 个进口药品和 9 个国产药品;其中包含化学药品 11 个、生物制品 13 个,中药 3 个。
笔者根据CDE公开的2020年度药品审评报告内容,整理了各类药品的上市后研究技术要求,并总结其中共性信息,供行业同仁参考。
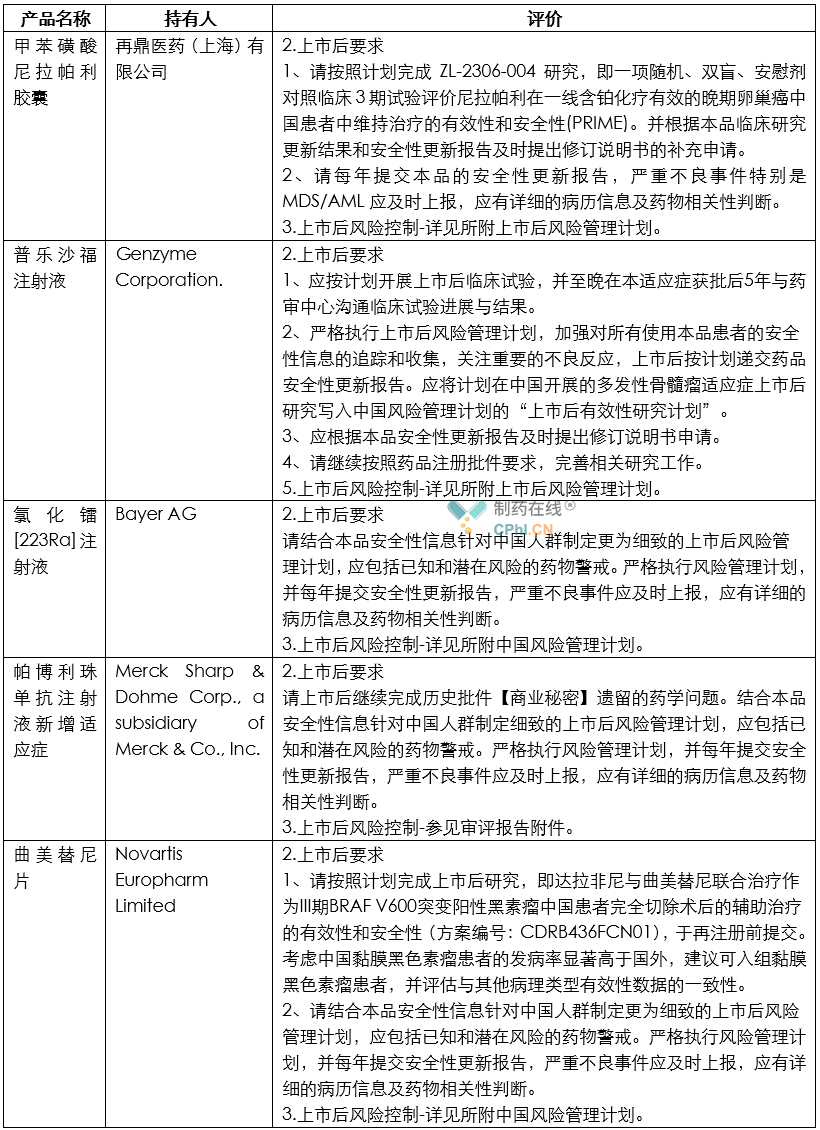
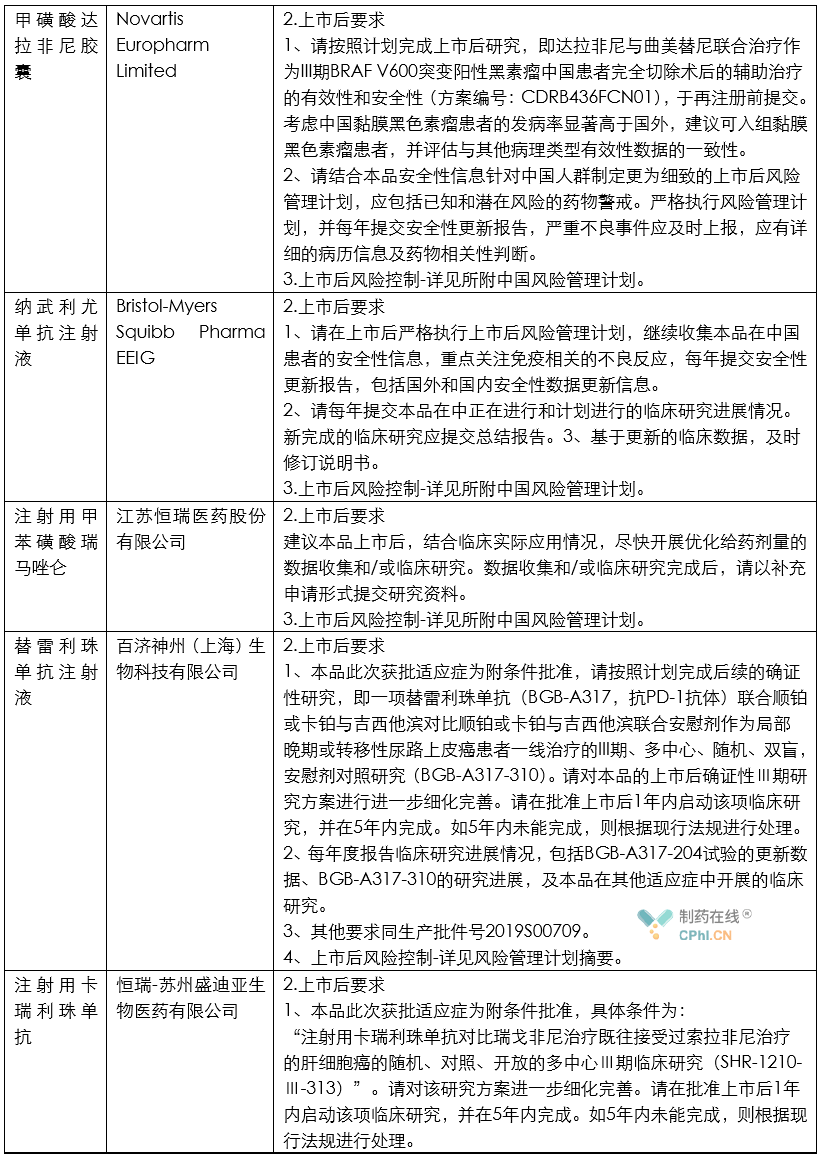
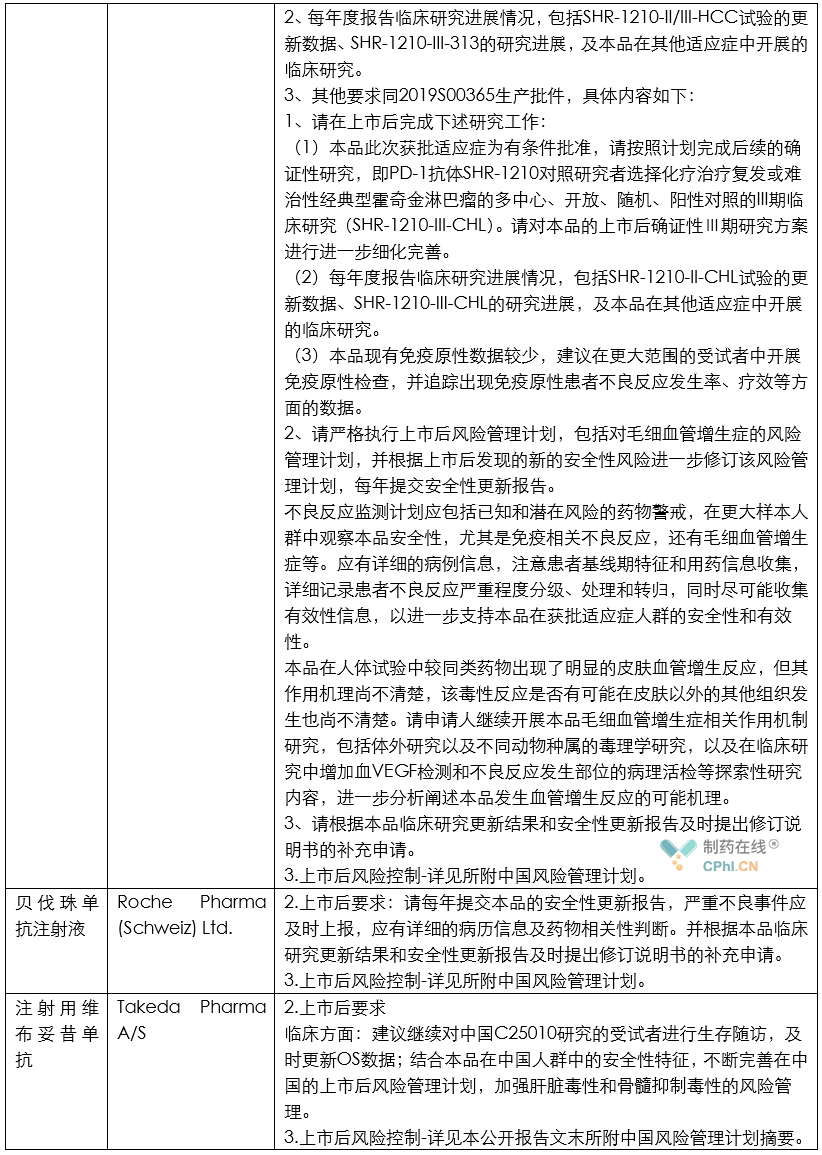
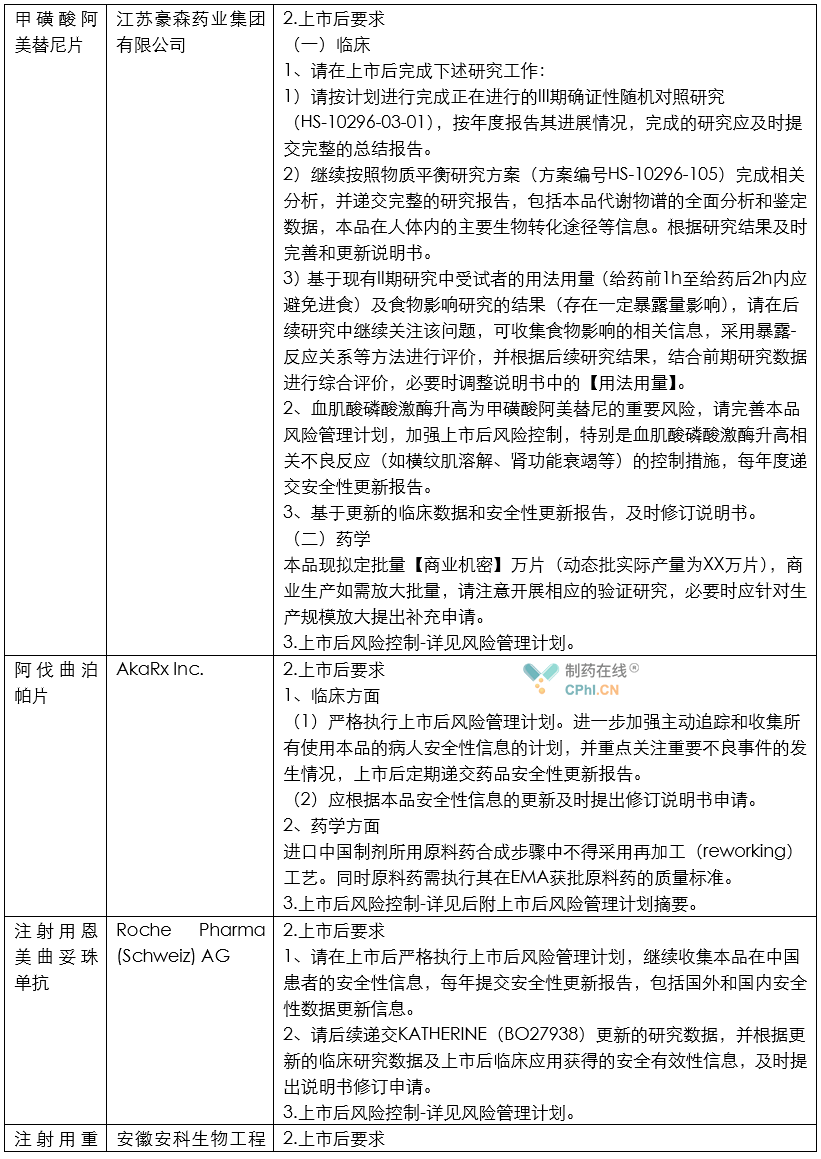
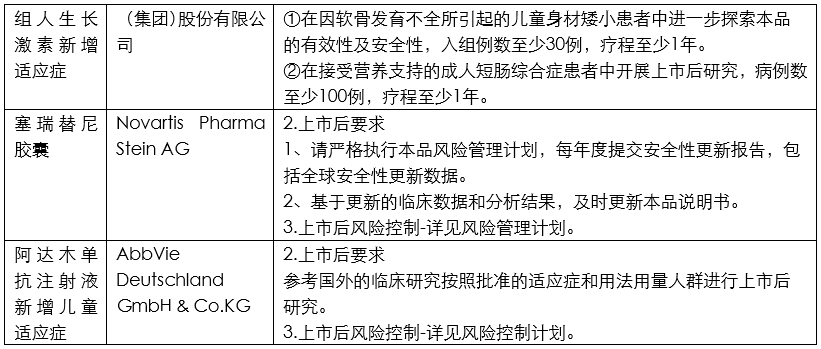
分析
根据上面列举和汇总的这些创新药和改良型创新药的审评报告所提示的信息,目前药政机构对于获批上市药品给出的上市后研究任务一般包括如下内容:
第一,上市后风险控制计划是常规要求。目前,虽然《药品管理法》和《药品注册管理办法》虽然提到MAH需要针对药品建立上市后风险控制计划;但是从目前情况看,这项要求主要针对创新药和改良型创新药提出类似要求。
第二,部分药品虽然获得批准,但是由于临床试验期间被发现存在明显不良反应,因此官方提出药品上市后要继续对相关不良反应进行深入研究。例如恒瑞制药的注射用卡瑞利珠单抗被发现存在明显毛血管增生的不良反应,因此恒瑞制药需要:严格执行上市后风险管理计划,包括对毛细血管增生症的风险管理计划,并根据上市后发现的新的安全性风险进一步修订该风险管理计划,每年提交安全性更新报告。 不良反应监测计划应包括已知和潜在风险的药物警戒,在更大样本人群中观察本品安全性,尤其是免疫相关不良反应,还有毛细血管增生症等。应有详细的病例信息,注意患者基线期特征和用药信息收集,详细记录患者不良反应严重程度分级、处理和转归,同时尽可能收集有效性信息,以进一步支持本品在获批适应症人群的安全性和有效性。
第三,部分依赖境外人种数据上市的药品,需要针对中国患者搜集和补充不良反应数据,并完善说明书信息等。例如,Bristol-Myers Squibb Pharma EEIG 公司的纳武利尤单抗注射液获批上市后,被要求:继续收集本品在中国患者的安全性信息,重点关注免疫相关的不良反应,每年提交安全性更新报告,包括国外和国内安全性数据更新信息。
第四,部分产品虽然获得批准,中国官方担心MAH为了降低成本,随意变更工艺,因此对API的工艺和质量标准都提出了要求。例如阿伐曲泊帕片获得批准后,官方提出的要求是:进口中国制剂所用原料药合成步骤中不得采用再加工(reworking)工艺。同时原料药需执行其在EMA获批原料药的质量标准。
第五,部分产品虽然获得批准,但是部分临床数据还不充分,还需要继续补充支持适应症的临床数据。例如百济神州的替雷利珠单抗注射液虽然获得批准,但是官方审评结论是:本品此次获批适应症为附条件批准,请按照计划完成后续的确证性研究,即一项替雷利珠单抗(BGB-A317,抗PD-1抗体)联合顺铂或卡铂与吉西他滨对比顺铂或卡铂与吉西他滨联合安慰剂作为局部晚期或转移性尿路上皮癌患者一线治疗的III期、多中心、随机、双盲,安慰剂对照研究(BGB-A317-310)。请对本品的上市后确证性Ⅲ期研究方案进行进一步细化完善。请在批准上市后1年内启动该项临床研究,并在5年内完成。如5年内未能完成,则根据现行法规进行处理。
总之,随着药品审评审批制度的持续改革和完善,中国市场也会成为全球创新药上市的首选地;这一切既是机会,也对中国药品审评审批制度提出了挑战。希望中国NMPA可以持续改革和完善,促进中国医药上市规范、健康发展。
说明:本文不构成任何投资建议和参考。
参考文献
1- NMPA官网信息
2- CDE官网信息
作者简介:zhulikou431,高级工程师、PDA会员、ISPE会员、ECA会员、PQRI会员、资深无菌GMP专家,在无菌工艺开发和验证、药品研发和注册、CTD文件撰写和审核、法规审计、国际认证、国际注册、质量体系建设与维护领域,以及无菌检验、环境监控等领域皆具有较深造诣。近几年开始着力关注制药宏观领域趋势分析和制药企业并购项目的风险管理工作。




合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57





















