近期,很多公众号和网站宣传:中国药政官方将推出2022年版《药品生产质量管理规范实施指南》,并且开展了预订活动。消息一出,立即引发行业持续关注。
2011年3月1日生效的《药品生产质量管理规范》(2010年修订)距今确实过去十多年了。在这过去的岁月中,中国制药行业发生了巨大变化;不管是监管法规体系,还是制药行业产品布局和行业态势都和过去显著不同了。
在客观形势发生巨大变化的情况下,修订法规和配套文件是合适的操作。例如2020年4月国家局就修订了GMP附录3生物制品和附录4血液制品,这就是很好的做法。对比来看,目前这种不修订2010版GMP规范却修订出版2022版GMP实施指南的操作有些类似舍本逐末,不分主次。
笔者不揣冒昧,结合过去多年对2010版GMP和附录进行研究的心得,对相关不合适的条款提出建议,希望为医药行业提供参考和借鉴。
第一部分:2010版GMP通则部分修订展望
修订建议1-GMP文件应该增加文件历史和编号
尽管药监局检查制药企业时,都要求制药企业建立完善的文件体系,但是我们无奈的看到,药监局自身法规文件体系的不完善,是显而易见的。
在2011年3月1日发布2010版GMP时,附录1-附录5的编号还是清晰的,后来的相关附录编号就很难搞清了,不利于法规学习和普及。
另外,看看下面的例子:
下面截图来自ICH Q3D(R1)的部分内容,如果国内有一个法规文件的历史说的可以这样清晰,对于法规维护人员和学习人员,都是福音。
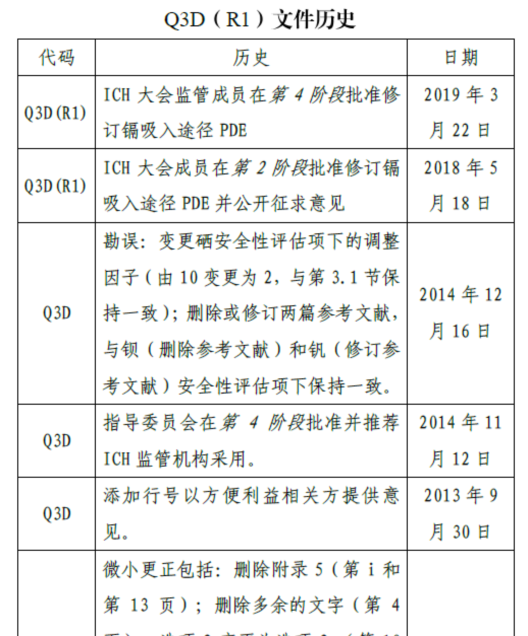
再看看下面截图

如果国内指导原则或者指南,都有类似的清晰编号体系,对于法规指南维护人员和学习人员,都是福音。
修订建议2-GMP通则部分语句需要修改
例如,中国GMP2010版通则的:
第二百四十四条 改变原辅料、与药品直接接触的包装材料、生产工艺、主要生产设备以及其他影响药品质量的主要因素时,还应当对变更实施后最初至少三个批次的药品质量进行评估。如果变更可能影响药品的有效期,则质量评估还应当包括对变更实施后生产的药品进行稳定性考察。
解析:上面三个批次的要求是不合适的。因为根据《药品上市后变更管理办法(试行)》和配套的已上市化学/中药/生物制品药学变更技术指导原则,药品注册事项变更分为微小变更、中等变更和重大变更。不是所有变更都需要在变更后进行三个批次评估的。
第二部分:2010版GMP附录部分修订展望
修订建议3-中国GMP2010版附录1无菌药品的部分条款需要修订
第九条 无菌药品生产所需的洁净区可分为以下4个级别:
。。。
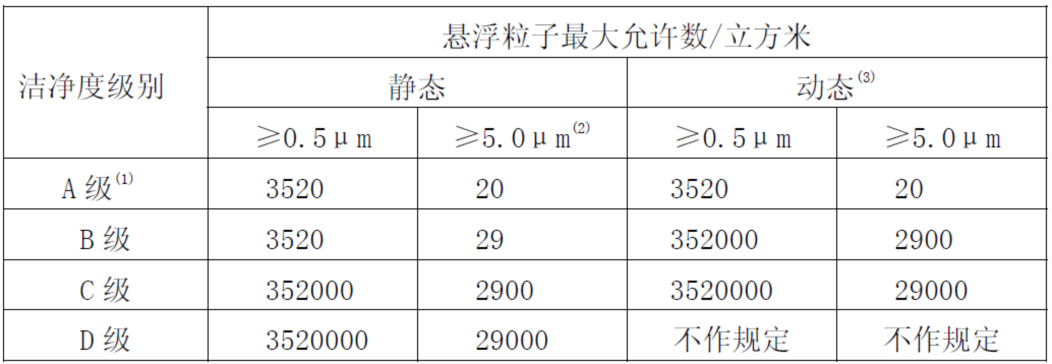
注:
(1)为确认A级洁净区的级别,每个采样点的采样量不得少于1立方米。A级洁净区空气悬浮粒子的级别为ISO 4.8,以≥5.0μm的悬浮粒子为限度标准。B级洁净区(静态)的空气悬浮粒子的级别为ISO 5,同时包括表中两种粒径的悬浮粒子。
说明:估计上面这段注释汇总的涂红内容,大部分药学技术人员读的别扭。如果你阅读发现别扭,那就对了,这是因为这段文字来自EU GMP附录1的翻译,而且翻译的不通顺,因此影响了阅读和理解。
比较通顺的翻译是:当以≥5.0μm的悬浮粒子为限度标准时,A级洁净区悬浮粒子的级别为ISO 4.8级。
我们仔细阅读上面的图表和注释,就明白了:
---判断洁净区级别,需要采用0.5um粒子和5um粒子来综合判断。
---对于B级区域,静态时,不管是0.5um粒子还是5um粒子,都是符合ISO14644-1标准中的5级标准的。
---对于A级区域,静态和动态标准一致;其中,如果按照0.5un粒子来判断,是符合ISO14644-1标准的5级标准的。但是如果按照5um粒子来判断,不符合ISO14644-1标准的5级标准,而是符合ISO14644-1标准的4.8级标准。
---上面翻译的错误在于,违背了中文语言习惯。中文语言习惯是定语和状语在前,主语在后;而英语正好相反。
修订建议4-中国GMP2010版附录2原料药部分条款需要修订
第三十七条 重新加工:
(一)应当对重新加工的批次进行评估、检验及必要的稳定性考察,并有完整的文件和记录,证明重新加工后的产品与原工艺生产的产品质量相同。可采用同步验证的方式确定重新加工的操作规程和预期结果。
说明:上面这段红色文字存在合理性问题。任何单位不可能采用同步验证的方式来确定重新加工工艺。
因为:
---重新加工面对的批次肯定是失败批次,这样的批次发生是偶然的。任何单位不会故意制造失败,除非是自虐式操作。
---如果选择自然的失败批次,现实情况是:这样失败的批次往往是偶发的,第一批失败批次和第二批失败批次之间不知间隔多长时间?另外,也没有人可以确保第一批失败批次和第二批失败批次发生的原因相同。如果真有这样的人,应该是阿拉伯的先知。看看下面这抗日神剧中的台词,就明白这样逻辑是多么荒唐!

修订建议5-中国GMP2010版附录2原料药部分条款需要修订
第三十三条 污染的控制:
(一)同一中间产品或原料药的残留物带入后续数个批次中的,应当严格控制。带入的残留物不得引入降解物或微生物污染,也不得对原料药的杂质分布产生不利影响。
说明:上面这段文字因为没有准确翻译ICH Q7指南的内容,导致意思偏移;并进而导致很多中国GMP检查人员在检查中国企业时,提出不合理要求。那就是,在原料药企业同品种不同批次生产时,要求每批次生产后,必须彻底清洁,不允许有明显残留物进入下一批次,否则视为混批。
ICH Q7原文和翻译如下:
8.50 Residual materials can be carried over into successive batches of the same intermediate or API if there is adequate control. Examples include residue adhering to the wall of a micronizer, residual layer of damp crystals remaining in
a centrifuge bowl after discharge, and incomplete discharge of fluids or crystals from a processing vessel upon transfer of the material to the next step in the process. Such carryover should not result in the carryover of degradants or microbial contamination that may adversely alter the established API impurity profile.
翻译:如果有足够的控制措施,相同的中间体或者API的残留物料可以被携带进入后续连续批次。这样的例子包括:附着在微粉机器壁上面的残留物料、离心机卸料后残留在内部器壁上面的潮湿晶体,和将物料转移到工艺中的下一步骤时,由于卸料不完全而导致的流体物料或晶体物料在处理容器中的残留。这样的残留物不应导致降解物残留或微生物污染成可能对已建立的API杂质档案产生不利影响的因素。
解析:如果仔细阅读ICH Q7上述的指南原文和翻译文本,你会发现,ICH Q7指南原文的意思是,如果API制造商采取了足够控制措施和进行了评估,在生产同一种API/中间体时,可以连续生产多个批次后进行一次彻底清洁;而不是每生产完毕一个批次,就进行彻底清洁。否则,连续制造在中国永远只能是空想。
修订建议6-动物源性材料控制力度不足
2020年4月23日,NMPA发布58号公告,发布修订后的GMP附录《生物制品》。但是其中部分内容不合适。
第四十七条 用于生产的培养基/培养液应与批准的一致;培养基应进行适用性检查;禁止使用来自牛海绵状脑病疫区的牛源性材料,并应符合《中华人民共和国药典》的相关要求。
解析:上述红色文字不合适。建议修订为:禁止使用来自疫区的相应动物源性材料。
参考文献
1-《药品生产质量管理规范》(2010年修订)通则和附录
2-《药品上市后变更管理办法(试行)》
3-《已上市化学药品药学变更研究技术指导原则(试行)》
4-《已上市中药药学变更研究技术指导原则(试行)》
5-《已上市生物制品药学变更研究技术指导原则(试行)》
作者简介:zhulikou431,高级工程师、PDA会员、ISPE会员、ECA会员、PQRI会员、资深无菌GMP专家,在无菌工艺开发和验证、药品研发和注册、CTD文件撰写和审核、法规审计、国际认证、国际注册、质量体系建设与维护领域,以及无菌检验、环境监控等领域皆具有较深造诣。近几年开始着力关注制药宏观领域趋势分析和制药企业并购项目的风险管理工作。




合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57





















