在2019年下半年,随着《药品管理法》(2019年修订版)颁布以后,制药企业技术人员学习任务明显加重。在国庆节前夕,国家局发布了三个重要法规的征求意见稿,分别是《药品注册管理办法》、《药品生产监督管理办法》和《药品经营监督管理办法》。10月15日,国家局又发布了《国家药监局综合司公开征求《化学药品注射剂仿制药质量和疗效一致性评价技术要求(征求意见稿)》《已上市化学药品注射剂仿制药质量和疗效一致性评价申报资料要求(征求意见稿)》意见》。
关于注射剂一致性评价的法规,早在2017年12月22日,国家局审评中心(cde)发布了《关于公开征求《已上市化学仿制药(注射剂)一致性评价技术要求》意见的通知》。但是这份征求意见稿在近两年的时间内,一直没有进一步修订完善的消息。但是,中国制药行业中一些优秀企业早已经开始着手启动注射剂产品的一致性评价工作,例如山东齐鲁制药和四川汇宇制药等公司。
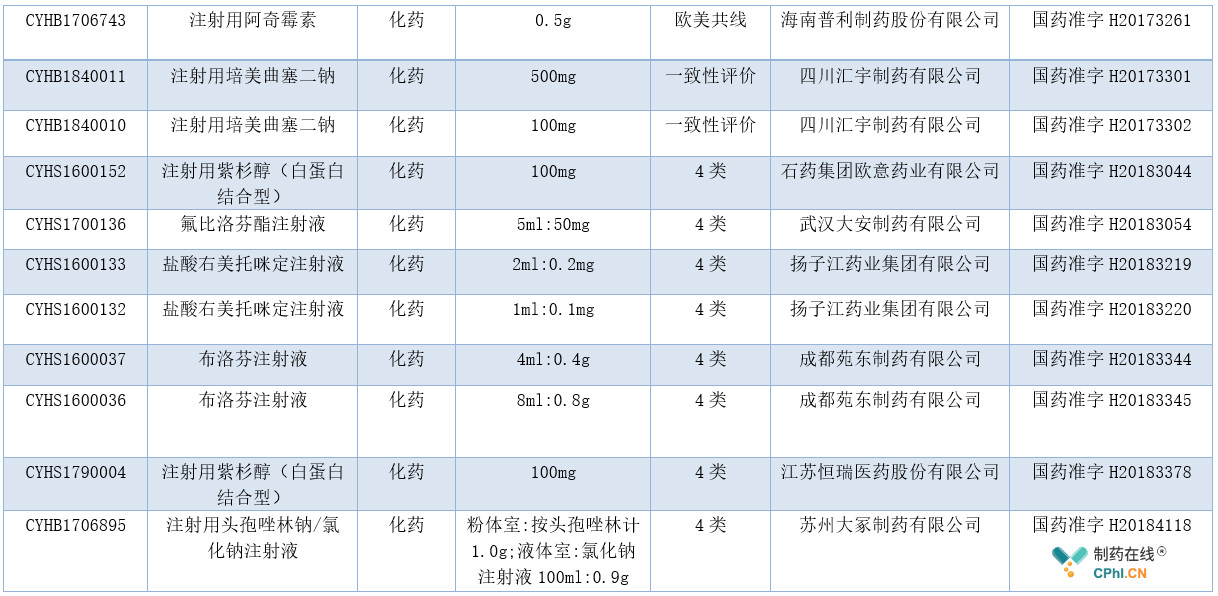
截止到2018年12月底为止,据不完全统计,通过一致性评价的药品情况参见下表:
不仅如此,国家局在陆续发布的化学仿制药参比制剂目录的文件中也不断增加无菌制剂(含滴眼剂和注射剂)的品种。例如,2019年9月16日,国家局发布的第24批化学仿制药参比制剂目录中就包含多个无菌制剂的RLD;这份最新的RLD目录包括贝美素噻吗洛尔滴眼液、醋酸泼尼松龙滴眼液和间苯三酚注射液等多个无菌制剂品种。
根据上面这些政策信号,并结合昨天国家局发布的最新注射剂一致性评价相关文件,可以预见的是,在2019年或者2020年初,注射剂一致性评价将会全面推行。
下面笔者结合昨天刚发布的《化学药品注射剂仿制药质量和疗效一致性评价技术要求(征求意见稿)》和《已上市化学药品注射剂仿制药质量和疗效一致性评价申报资料要求(征求意见稿)》,解析要点,并提供自己的建议,期望可以为行业从业者的应对有所帮助。
要点1:申报人必须对所申报产品具有全面了解
这份最新的技术要求征求意见稿对于申报人的战略视野要求较高。原文是:申请人应全面了解已上市注射剂的国内外上市背景、安全性和有效性数据、上市后不良反应监测情况,评价和确认其临床价值。应该说,这份要求在目前研发深入全面推进的历史背景下是合适的。一个制药公司的项目负责人,要想成功推动一个项目进行并成功申报,必须具有丰富的专业知识、对法规精准掌握、对于研发和工业结合把握得当,并掌控合适的公司内部资源和外部资源。可以说,如果一个申报人不能对要负责的项目有全面理解和掌握,所提交的申报资料质量是会让人担忧的。官方提出这样的要求是合情合理的。
要点2:RLD选择原则明确
在过去,国家药监局发布了口服固体制剂一致性评价的RLD选择原则,目前根据这份技术要求,这些原则也是适用于注射剂一致性评价工作的:申请人应按照国家局发布的《化学仿制药参比制剂遴选与确定程序》选择参比制剂,参照本技术要求和国内外相关技术指导原则开展一致性评价研究工作。我们上面也提到,在最新一期的官方发布的RLD目录中,已经大规模出现无菌制剂(含滴眼剂、注射剂品种)的目录。
要点3:文献检索在处方和工艺确定过程中发挥关键作用
和口服固体制剂不同,小分子化学药品的简单剂型一般是不需要进行临床试验的。在这种情况下,为了确保注射剂仿制药和原研药品具有高度一致的质量效果和质量属性,就需要遵循国际上通行的Q1/Q2原则。所谓Q1/Q2原则指的是化学药注射剂仿制药需要在原辅料品种上面和原研保持一致,在原辅料的投料数量方面和原研尽量保持一致。如果要做到这一点,就需要强化制药企业研发人员的信息检索能力。
例如,这份技术要求草案中提到如下细节:
※处方-注射剂中辅料种类和用量通常应与参比制剂(RLD)相同。辅料的用量相同是指仿制药辅料用量为参比制剂相应辅料用量的95%-105%。如附带专用溶剂,应与参比制剂的专用溶剂处方一致。
※申请人可以提交与参比制剂抑菌剂、缓冲剂、pH调节剂、抗氧剂、金属离子络合剂不同的处方,但需标注不同之处,阐述选择的理由,并研究证明上述不同不影响所申请产品的安全性和有效性。
※辅料的浓度或用量需符合FDA IID数据库限度要求,或提供充分依据。过量投料建议参考ICH Q8相关要求。
要点4:无菌工艺和灭菌工艺研究水平必须符合目前行业最新要求
无菌产品可以采用最终灭菌工艺来生产,也可以采用非最终灭菌工艺来生产。但是不管是哪种生产工艺,所得最后无菌产品的无菌保证水平必须符合最新的法规要求和行业共识。
这份技术要求草案提到如下细节:(1)为了有效控制热原(细菌内毒素),需加强对原辅包、生产过程等的控制,注射剂生产中不建议使用活性炭。(2)根据生产工艺进行过滤器相容性研究。根据溶液的特点和生产工艺进行硅胶管等直接接触药液容器的相容性研究。(3)如参比制剂存在过量灌装,仿制药的过量灌装宜与参比制剂保持一致,如不一致需提供合理性论证。
对于中国制药企业,要想开发出符合目前国际制药行业共识和最新法规要求的灭菌工艺或者无菌工艺,需要学习如下指南:
※FDA无菌工艺指南
※日本PMDA无菌工艺指南
※EU GMP附录1和PIC/S无菌指南
※PDA 关于无菌工艺开发和研究的技术报告
※EMA关于灭菌工艺开发的指南
※USP/EP/JP关于灭菌工艺的相关要求
另外,中国制药企业如果要采用无菌工艺来生产无菌药品,必须高度重视环境监控问题(EM)。在2018年,中国山东某企业和广东某企业先后被欧美药政机构警告,都是因为EM存在缺陷所致。
要点5:各类无菌验证工作在注射剂一致性评价中处于重点地位
一个无菌药品要想实现质量属性稳定,除了扎实的研发工作,还需要在日后商业化生产中确保关键工艺参数和质量属性稳定,才能实现自己的产品质量设计期望。这其中,对于无菌产品而言,各类灭菌工艺验证工作显得尤其重要。因此,这份最新的技术要求草案提出如下细致要求:
(1)灭菌/无菌工艺验证
对于终端灭菌药品,至少进行并提交以下验证报告:
o 药品终端灭菌工艺验证;
o 直接接触药品的内包材的除热原验证或供应商出具的相关证明资料;
o 包装系统密封性验证,方法需经适当的验证;
o 保持时间(含化学和微生物)验证。
对于无菌灌装产品,至少进行并提交以下验证报告:
o 除菌工艺的细菌截留验证;
o 如不采用过滤除菌而采用其他方法灭菌,提供料液/大包装药的灭菌验证;
o 直接接触无菌物料和产品的容器密封系统的灭菌验证;
o 直接接触产品内包材的除热原验证或供应商出具的相关证明资料;
o 无菌工艺模拟试验验证,并明确试验失败后需要采取的措施;
o 包装系统密封性验证,方法需经适当的验证;
o 保持时间(含化学和微生物)验证。
(2)生产工艺验证
提供工艺验证资料,包括工艺验证方案和验证报告。
要点6:对于原辅包采取最严格要求
和口服固体制剂不同的是,化学药品注射剂仿制药要求尽量和原研药品保持相同的辅料构成和数量比例。不仅如此,而对原辅料的质量规格和安全属性提出更高要求。
对于原料药,技术要求草案提出:制剂生产商需结合原料药生产工艺,根据现有指导原则和相关文件(含国家局2008年7号文)对原料药的质量进行充分研究与评估,必要时修订有关物质检查方法,增加溶液澄清度与颜色、溶剂残留、细菌内毒素、微生物限度等检查,并提供相关的验证资料,以满足注射剂工艺和质量的控制要求;同时需关注对元素杂质和致突变杂质的研究和评估。制剂生产商需根据注射剂持续稳定生产的需要,对原料药来源和质量进行全面的审计和评估,在后续的商业化生产中保证供应链的稳定。如发生变更,需进行研究并按相关技术指导原则进行研究和申报。
对于辅料,技术要求草案提出:辅料应符合注射用要求,制定严格的内控标准。除特殊情况外,应符合现行中国药典要求。
可以预见的是,虽然在2015版药典实施后,中国辅料生产行业质量有所提高,但是大部分辅料还不是注射级或者无菌级的。因此都需要制剂商慎重选择供应商。也可以预见,那些未雨绸缪的领先辅料供应商,在未来市场竞争中处于较强的优势地位。
对于内包材,由于在生命周期内,内包材对于注射剂影响是非常直接和关键的,因此技术要求草案提出如下要求:注射剂使用的直接接触药品的包装材料和容器应符合国家局颁布的包材标准,或USP、EP、JP的要求。根据药品的特性和临床使用情况选择能保证药品质量的包装材料和容器。
按照《化学药品注射剂与塑料包装材料相容性研究技术指导原则(试行)》、《化学药品注射剂与药用玻璃包装容器相容性研究技术指导原则(试行)》、《化学药品与弹性体密封件相容性研究技术指导原则(试行)》等相关技术指导原则开展包装材料和容器的相容性研究。
根据加速试验和长期试验研究结果确定所采用的包装材料和容器的合理性,建议在稳定性考察过程中增加样品倒置等考察,以全面研究内容物与胶塞等密封组件的相容性。注射剂使用的包装材料和容器的质量和性能不得低于参比制剂,以保证药品质量与参比制剂一致。
从上面这些要求可以看出,注射剂仿制药生产商必须深入研究RLD使用的内包材,最好进行元素分析,以判断内包材对于产品的影响情况。
要点7:质量研究必须遵循ICH Q8指南思路
质量研究是所有药品开发阶段的重点工作。对于化学药注射剂一致性评价工作,这份文件最新提出ICHQ8所要求的研发路线:QTPP-CQA-CPP-CP。例如技术要求规定:根据目标产品的质量概况(QTPP)确立制剂的关键质量属性(CQA),通常注射剂的CQA包括但不限于以下研究:性状、鉴别、复溶时间、分散时间、粒径分布、复溶溶液性状、溶液澄清度、溶液颜色、渗透压/渗透压比、pH值/酸碱度、水分、装量、装量/重量差异、含量均匀度、可见异物、不溶性微粒、细菌内毒素、无菌、元素杂质、残留溶剂、有关物质(异构体)、原料药晶型/粒度、含量等。
注射剂是直接进入人体的药品,因此安全性一直是监管方和申报方所重点关注的。这份文件除了继续强调有关物质检查的必要性,还增加了对致突变杂质的要求。例如规定如下:根据相关文献、参比制剂的情况,通过对生产工艺、产品降解途径的分析,判断是否可能产生潜在的致突变杂质,必要时进行针对性的研究,根据研究结果按照相关技术指导原则进行控制。
应该说,虽然2018年中期发生的缬沙坦事件是口服固体产品事件,但是随着这个问题的影响扩大,监管方把监管视角延伸到注射剂方面也是非常正常的。一般国际上建议采用ICH M7指南来研究和控制基因**杂质。
另外,对于元素杂质要求,也是这份文件新特点:根据ICH Q3D的规定,通过科学和基于风险的评估来确定制剂中元素杂质的控制策略,包括原辅包、生产设备等可能引入的元素杂质。
要点8:和RLD对比要求比口服制剂要求更高
在一致性评价工作中,将自己产品和RLD对比是常规要求。但是对比程度,口服固体要求一般要求不高。从这份文件看,要求申报人将自己开发的产品全面和RLD对比,具体要求如下:自研产品应与参比制剂进行全面的质量对比(含杂质谱对比),保证自研产品与参比制剂质量一致。参比制剂原则上应提供多批次样品的考察数据,考察与一致性评价紧密相关的关键质量属性。
可以预见的是,RLD采购和标准品采购也会消耗很大部分项目资金。
要点9:稳定性研究增加较多新要求
在药品生命周期中,稳定性研究重要是毋庸置疑的。这份技术要求除了沿袭过去常见要求,还针对注射剂提点提出新要求:注射剂稳定性研究的加速试验、长期试验应在符合GMP条件下进行,可综合考虑申报注射剂产品的特点,如产品规格、容器、装量、原辅料浓度等,按照相关技术指导原则设计稳定性研究方案,考察在贮藏过程中易发生变化的,可能影响制剂质量、安全性和/或有效性的项目。若注射剂处方中含有抗氧剂、抑菌剂等辅料,在稳定性研究中还要考察这些辅料含量的变化情况。稳定性考察初期和末期进行无菌检查,其他时间点可采用包装系统密封性替代。包装系统密封性可采用物理完整测试方法(例如压力/真空衰减等)进行检测,并进行方法学验证。一般应提供不少于6个月的稳定性研究数据。根据稳定性研究结果,参考参比制剂信息确定贮藏条件,仿制药的稳定性应不低于参比制剂。
虽然上面一段的最后一句是合理的,但是从近几年审评情况看,很多仿制药产品的稳定性比RLD差也可以获得批准。这应该是采用了QRM原则来判断的合理原则。
另外,这份技术要求也提到,稳定性试验需要考察最差摆放位置的情况;这也是一个新变化。
要点10:高度关注特殊注射剂的质量熟悉和工艺特征相关性
对于一些特殊注射剂(如脂质体、静脉乳、微球、混悬型注射剂等),这份文件除了要求遵循常规技术要求外,还提出如下特定要求:
※处方工艺:处方原则上应与参比制剂一致,建议对辅料的型号及可能影响注射剂体内行为的辅料的CQA进行研究。特殊注射剂的生产工艺可能影响药物体内行为,需深入研究;对于采用无菌工艺生产的特殊注射剂,需特别注意各生产步骤的无菌保证措施和验证。注册批和商业批的生产工艺及批量原则上应保持一致。
※质量研究:考察的关键质量属性可能包括但不限于以下内容:理化性质(如性状、黏度,渗透压摩尔浓度,pH值/酸碱度等),Zeta电位,粒子形态,粒径及分布(如D10,D50,D90等),体外溶出/释放行为,游离和结合药物,药物晶型和结晶形态。原则上应提供至少3批次参比制剂样品的质量对比考察数据。
※BE/临床试验的考虑:应采用商业批量的样品进行BE试验和/或临床试验。对于FDA或EMA已公布指导原则的特定注射剂品种,建议参照其技术要求开展与参比制剂的对比研究。
要点11:对于说明书和质量标准都提出新要求
这份新文件,除了要求申报人符合中国最新法规外,还对说明书管理和质量标准管理提出最新要求:
※药品说明书的拟定:申请人需检索并追踪参比制剂说明书的变更情况,参考最新版参比制剂说明书,合理拟定一致性评价药品说明书。
※药品标准:药品注册标准收载检验项目少于药典规定或质量指标低于药典要求的,应执行药典规定。
要点12:无需开展一致性评价的品种有需要进行质量提升
对于很多成分简单,临床使用历史悠久的品种,这份最新文件没有要求进行一致性评价,但是仍然要求进行质量提升。例如规定如下:氯化钠注射液、葡萄糖注射液、葡萄糖氯化钠注射液、注射用水、部分**药物(如锝〔99mTc〕)等品种无需开展一致性评价,需进行质量提升研究,灭菌工艺、滤器与包材选择(含相容性研究)等应符合相关技术要求。
总结
基于上面的分析和讨论,我们建议制药企业做好如下工作:
---技术团队组建工作,要选择具有最新法规知识、熟悉工业生产和情报搜集人员。
---持续加强ICH 指南学习,例如ICH M7/ICH Q3/ICH Q1等指南。
---加快市场调查,评价投入和汇报是否平衡。
---加快联系外部单位,核实包材相容性和其他质量要求成本费用。
---加快联系RLD供应商,是否可以快速买到核实的参比药品。
作者简介:zhulikou431,高级工程师、PDA会员、ISPE会员、ECA会员、PQRI会员、资深无菌GMP专家,在无菌工艺开发和验证、药品研发和注册、CTD文件撰写和审核、法规审计、国际认证、国际注册、质量体系建设与维护领域,以及无菌检验、环境监控等领域皆具有较深造诣。近几年开始着力关注制药宏观领域趋势分析和制药企业并购项目的风险管理工作。




合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57





















