美国医药市场目前在全球具有地位,而管理美国医药市场秩序的美国FDA也以科学和规范被制药企业所认可。为了持续推进法规的有效实施,FDA在遵循相关法规的同时,发布很多配套技术指南,来向制药行业和相关机构展示自己的认知和理解;应该说,这些指南的发布和在相关行业的影响,对于促进监管方与被监管方之间的沟通,促进行业交流和促进共识,是非常有帮助的。
随着科学技术发展以及现实情况的变化,一些指南会变得和实际情况不一致;因此,FDA会每年启动指南起草和修订工作,以促进相关指南更新,向社会公众和制药行业展示自己的最新认知。在近期,FDA在官网发布了《Guidance Agenda ---New & Revised Draft Guidances---CDER Plans to Publish During Calendar Year 2020》,向各国制药企业介绍了在2020年度FDA关于技术指南起草和修订的具体计划。
第一部分:文件整体介绍
在文件开始,这份工作计划介绍了文件发布的依据是:Good Guidance Practices (GGPs) 或者21 CFR 10.115。而在后面的正文部分,这份工作计划告诉我们,在2020年度FDA新起草和修订的技术指南将覆盖14个领域。具体情况参见下表:
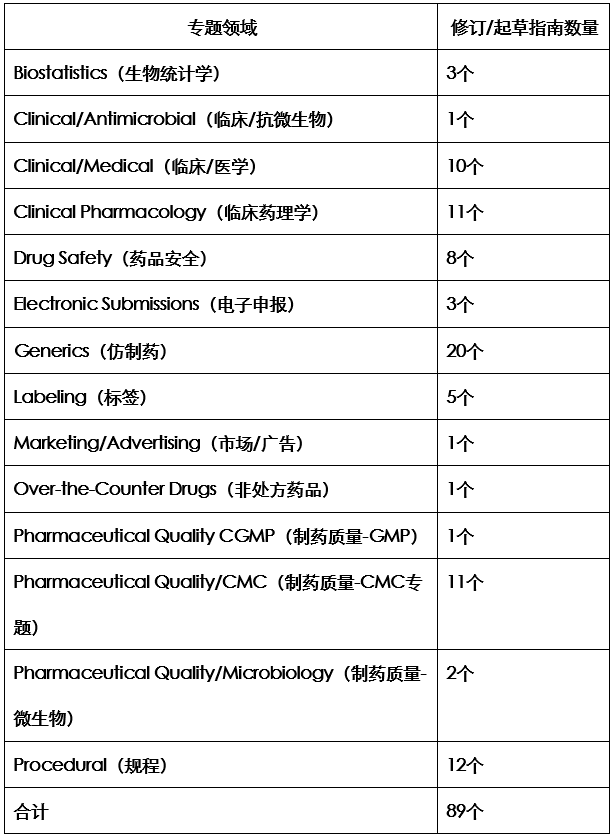
解析:从上面表格可以看出,如果在2020年完成这份工作计划,FDA需要在每个月完成7份多指南,任务不轻。而在制药行业关注的仿制药专题,和往年类似,在2020年度仍然安排了很多指南修订,显示了法规方对此领域的关注度持续不减。
第二部分:关键指南介绍
根据笔者的工作经验和这些指南对于中国制药行业的影响,选择关键指南介绍如下:
2.1-生物统计学(Biostatistics)
在这个领域,建议制药同仁关注指南《Multiple Endpoints in Clinical Trials》。这个指南介绍了当一个设计的临床试验有多个终点,最后试验数据如何处理和判断的建议。
2.2-临床药理学(Clinical Pharmacology)
在这个领域,建议关注《Dose Selection in Drug Development》。这个指南介绍了药品研发初期的剂量选择原则,体现了FDA对此问题的最新看法,对于持续高涨的中国创新药领域,会有很大帮助。
另外,建议关注《General Clinical Pharmacology Considerations for Pediatric Studies for Drugs and Biological Products》。这个指南介绍了在开发儿科药物时,需要关注的药理学问题。对于中国国家局持续推动的儿科用药开发,会有较大支持作用。
2.3-仿制药(Generics)
仿制药领域既是美国FDA关注的重点,也是中国国家药监局工作重点之一。这些新指南的更新,对于积极开展的仿制药一致性评价工作应该会有很多支持作用。
第一个需要关注的指南《180-Day Exclusivity: Questions and Answers; Revised Draft》。180-Day Exclusivity指的是第一个开发仿制药的企业可以根据Waxman-Hatch法案获得180天的独占期。原来FDA针对此问题发布过指南,本次修订会针对行业有争议问题进行澄清。
第二个需要关注的指南《ANDA Submissions - Refuse-to-Receive for DMF Facilities Deficiencies》。美国DMF指南最初版本是1989年版,在时隔30年后的2019年发布了修订版。针对制药企业递交过程中常犯错误,发布这个指南,可以提醒申报人员哪些错误不能犯。
第三个需要关注的指南是《ANDA Submissions - Refuse-to-Receive Standards: Questions and Answers》。美国针对仿制药申报,起草了RTR指南,翻译成汉语就是拒绝接受。中国药政部门在过去多年也发布了很多类似规定,行业内称之为"枪毙性条款"。2020年度FDA发布的这份指南应该对仿制药申报中的提醒作用更强。
第四个需要关注的指南是《Evaluation of Therapeutic Equivalence》。为什么要关注这份指南?因为中国药监局启动并推行的仿制药一致性评价工作,严格的讲,名称是不对的。国内称之为质量与疗效一致性评价,也是不科学的;规范称呼应该是疗效一致性评价。从来没有法规和理由来要求仿制药和参比药品(RLD)必须保持质量一致。在2018年中期发生的浙江华海缬沙坦时间,以及在2018年底发生的华北制药阿莫西林胶囊事件都证明了这一点。期待FDA这个指南可以解释清楚TE的要求,并对中国已经开展的口服固态药物的一致性工作和正在启动开展的注射剂一致性工作发挥作用。
第五个需要关注的指南是《Orange Book - Questions and Answers》。美国FDA的OB(橙皮书)不仅对美国国内仿制药研发发挥关键作用,对于中国仿制药工作也具有指导意义。期待这份指南可以帮助我们正确理解OB的使用要求。
第六个需要关注的指南是《Physico-Structural (Q3) Characterization of Topical Dermatological Drug Products Submitted in ANDAs》。中国正在开展的注射剂一致性评价,在国内指导原则上面,已经体现了FDA指南提到的Q1/Q2原则。但是对于一些复杂注射剂,如何理解Q3,如何正确应用,应该还是存在争议的。应该说,这份指南对于注射剂仿制药开发有较大指导意义。
第七个需要关注的指南《Statistical Approaches to Establishing Bioequivalence》。针对BE试验中使用的分析方法,FDA官网本来有声明:FDA不指定统计方法和工具,只要符合统计学原则即可。这个指南,应该对于仿制药行业开展BE试验的数据统计,产生很大影响和指导作用。
2.4-制药质量-GMP(Pharmaceutical Quality CGMP)
针对这个专题,建议关注《PET Drugs - Current Good Manufacturing Practice (CGMP); Revised Draft》。这个指南介绍了FDA对PET(正电子产品)的GMP期望和要求。
2.5-制药质量/CMC(Pharmaceutical Quality/CMC)
这部分内容涉及药品开发研究的药学内容,一致被国内药学工作者所关注,应该对中国官方监管态度也会产生影响。
第一个需要关注的指南是《ANDAs: Stability Testing of Drug Substances and Products Questions and Answers》。FDA曾经发布过针对仿制药的稳定性试验指南和相关问答,本次再次修订相关问题,应该可以促进稳定性技术问题的澄清和规范。
第二个需要关注的指南是《ICH Q12, General Considerations for FDA Implementation》。ICH Q12指南的主题是药品生命周期管理中的技术和注册考量,应该说对于上市后药品的管理,给出了很好的建议。不同药政当局应该参考ICH Q12,结合本国现实情况进行本国上市后药品的有效监管。这个指南体现了FDA对ICH Q12的最新认知,对于中国药政当局管理上市后药品,也是一个思路。
第三个需要关注的指南是《Inspection of Injectable Products for Visible Particulates》。注射剂产品的可见异物检查在不同国家存在争议和不同认识。这份指南会对意图进入美国市场的中国注射剂企业产生很大影响。
第四个需要关注的指南是《Setting Endotoxin Limits During Development of Investigational Oncology Drugs and Biologics》。这份指南本来是2019年度工作计划的一部分,由于过去一年没有完成,依次排到的2020年。应该说,严格的讲,中国药典针对内毒素限度计算公式是错误的。这个问题很早就被FDA和EU官方发现。FDA也一直想开发新的指南来帮助制药企业纠正这个问题。期待这个闹心的问题可以在2020年度得到解决。
2.6-制药质量-微生物(Pharmaceutical Quality/Microbiology)
在这个技术专题,建议关注指南《Microbiological Quality Considerations in Non-Sterile Drug Product Manufacturing》。应该说,随着中国药监局发布文件要求采用ICH Q6A指南,口服固态药品不需要再例行测试微生物项目。企业需要给予QRM来决定是否检测这个项目。但是是否对于口服固态制剂都可以取消此项目,应该说,FDA的这个新指南会给我们以帮助。
第三部分:影响分析
根据上面的信息汇总和分析,应该说,FDA2020年技术指南修订工作计划会对中国制药企业产生如下影响:
3.1-对于一致性评价工作产生支持作用
中国国家药监局已经启动口服固态制剂一致性评价工作,也在2019年底启动了注射剂一致性评价工作。应该说,尽管工作推进到一个新阶段,但是还是存在很多技术争议问题。经过上面的分析,FDA相关指南可以对中国境内仿制药工作产生一定的支持作用。
3.2-对于目标是美国市场的仿制药公司,产生持续影响
根据上面分析,可以看出2020年的FDA指南计划,对于各个方面都有更新和修订。这些新的技术要求,对于刚刚启动的项目、正在推进的研发项目和已经递交的项目都会产生影响。需要相关企业持续关注,早做应对措施。
3.3-提醒中国药政当局,对于ICH指南的采用不是简单发文就可以解决的
目前,中国国家药监局分别发布三个公告来推行ICH E/S/Q的专题指南。这项工作积极推进没有问题,但是官方必须考虑,这些ICH指南的内容和中国现有法规体系是冲突的,不是完全契合的。如果企业遇到类似问题,如何解决,这是不可回避的问题。
因此,建议中国国家药监局在相关外国指南采用过程中,必须充分设置征求意见阶段和主动邀请行业骨干企业和相关协会来讨论此问题。避免草率发文,导致企业莫衷一是的困局。
参考文献
1- 美国FDA2020年度指南起草和修订计划
2- 美国FDA官网信息
3- 《美国药品申报与法规管理》第一版
作者简介:zhulikou431,高级工程师、PDA会员、ISPE会员、ECA会员、PQRI会员、资深无菌GMP专家,在无菌工艺开发和验证、药品研发和注册、CTD文件撰写和审核、法规审计、国际认证、国际注册、质量体系建设与维护领域,以及无菌检验、环境监控等领域皆具有较深造诣。近几年开始着力关注制药宏观领域趋势分析和制药企业并购项目的风险管理工作。
点击下图进行CPhI & P-MEC China 2020观众预登记抽奖,奖品多多!





合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57





















