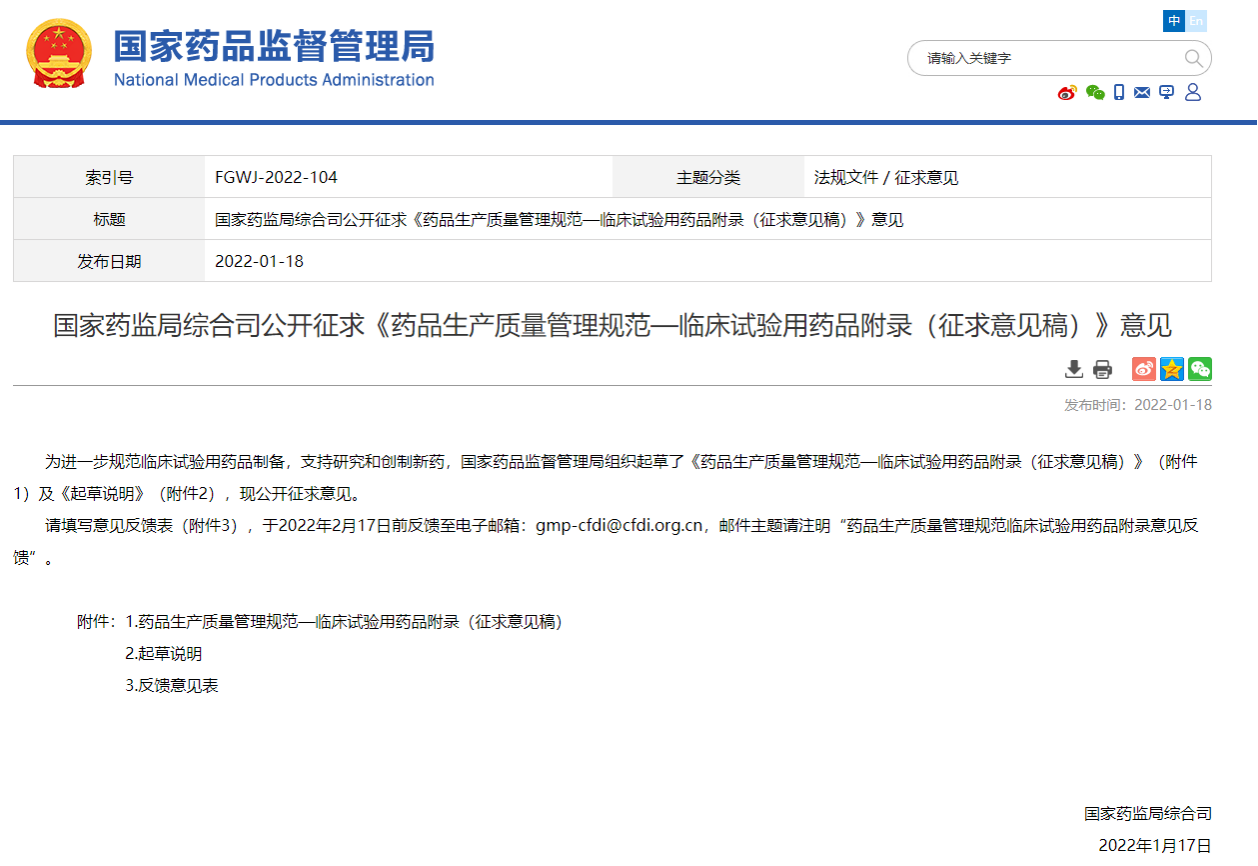
2022年1月18日,国家药品监督管理局官网重磅发布《药品生产质量管理规范-临床试验用药品附录》(征求意见稿)(成文日期:2022年1月17日),征求意见截止日期2022年2月17日,这是标志国家局把临床试验用药品纳入GMP附录,弥补了我国对临床试验用药品生产和质量检查在法规层面和技术层面的空白,进一步完善临床试验用药品监管长效机制。本文对《GMP附录-临床试验用药品》中关键点进行了分析。
一、为什么要单独起草《药品生产质量管理规范-临床试验用药品附录》
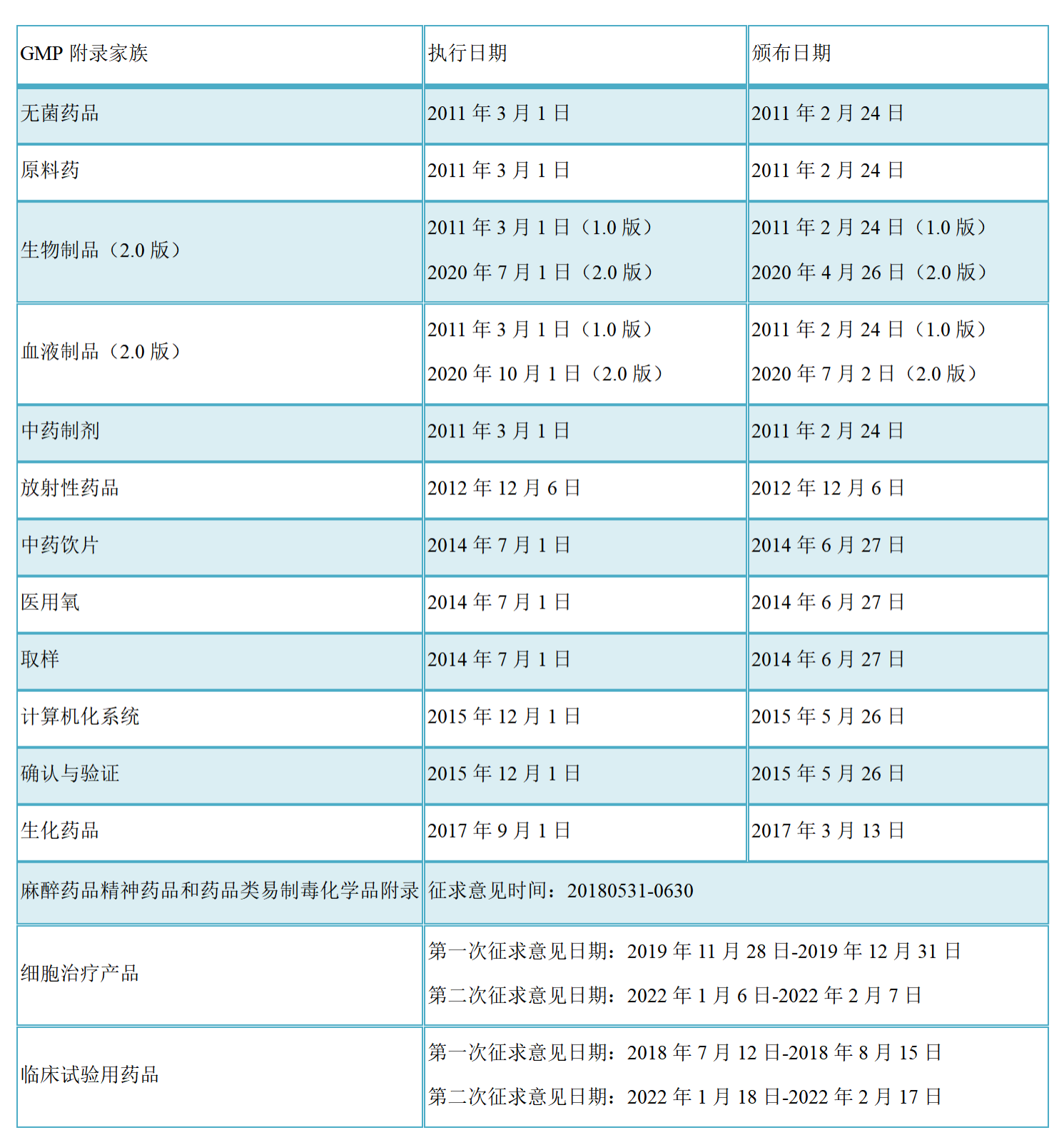
临床试验用药品有其特殊性,质量影响受试者安全和药物临床试验结果,其制备的质量管理应当遵循GMP相关基本原则。2018年7月国家药监局曾组织起草了《临床试验用药物生产质量管理规范(征求意见稿)》并公开征求意见,2022年1月18日,国家药品监督管理局官网重磅发布《药品生产质量管理规范-临床试验用药品附录》(征求意见稿),在2018年《临床试验用药物生产质量管理规范(征求意见稿)》基础上吸纳有关意见和建议,并结合当前法规、国际有关GMP最新修订进展情况进行了修改完善,我们将迎来国内首部《药品生产质量管理规范-临床试验用药品附录》,也是GMP附录第15个成员。
二、《GMP附录-临床试验用药品》关键点抢先看
2.1 适用范围是什么?

2.2 临床试验用药品人员有哪些特殊考虑?

2.3 临床试验用药品厂房与设施有哪些特殊考虑?

2.4 临床试验用药品物料有哪些特殊考虑?

2.5 临床试验用药品文件管理有哪些特殊考虑?

2.6 临床试验用药品制备管理有哪些特殊考虑?

2.7 临床试验用药品质量控制有哪些特殊考虑?

2.8 临床试验用药品批放行有哪些特殊考虑?

2.9 临床试验用药品发运有哪些特殊考虑?

三、小结
临床试验用药品的制备、使用和管理是药物临床试验的重要环节。2020版《药品注册管理办法》明确临床试验用药物应当在符合《药品生产质量管理规范》的车间制备,制备过程应当严格执行《药品生产质量管理规范》的要求,申请人对临床试验用药物的质量负责,临床试验用药品的质量与稳定性直接决定着临床试验结果的可靠性。为规范和指导临床试验用药品制备,支持研究和创制新药,国家药监局组织核查中心总结既往有关工作实践,参考相关国际做法,起草了《药品生产质量管理规范—临床试验用药品附录(征求意见稿)》。期待早日落地实施,该附录以GMP基本要求为基础,体现了临床试验阶段的特殊性,旨在最 大限度降低制备环节引入的风险,确保临床试验用药品质量,保障受试者的安全。
参考文献
[1]NMPA
作者简介:滴水司南,男,生物医药高级工程师,立足于生物医药行业质量管理工作,专注于生物医药产业。



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57






















