近期,CDE一纸新规落地,生物类似药说明书修订迎来关键转折!国家药品监督管理局药品审评中心(CDE)正式发布《生物类似药说明书撰写技术指导原则》(2025年第12号),不仅填补了行业规范空白,更对药企提出了“硬性合规”要求——已上市产品需按新规全面修订说明书,否则可能面临发补、退审甚至市场风险!新规重点划在哪?企业如何高效“避坑”?相较于旧版指南,此次更新在“适应症外推依据”“免疫原性数据呈现”等核心板块增设了更严苛的细则,稍有不慎就可能踩中CDE审评雷区。本文基于新规的核心变化,系统梳理新增条款的技术要点,结合行业实践案例,解析药企在自查与修订过程中需关注的风险节点与策略选择,以期为提升说明书科学价值提供可操作性建议。
一、生物类似药说明书新规核心变化内容差距分析
新《指导原则》的核心变化聚焦于以下几个方向:一是强化关键信息的科学呈现,明确要求说明书需清晰标注生物类似药与参照药的差异(如药学特征、适应症外推依据等),并细化免疫原性风险评估的披露要求;二是动态更新机制,强调上市后需持续追踪安全性数据并修订说明书,确保信息时效性。对于已上市生物类似药企业,开展差距分析需重点比对现有说明书与新规要求的差异。通过速查新规核心变化,药企可规避合规风险,同时提升说明书在临床实践中的实用性与可信度,为患者安全用药筑牢信息屏障。
1.生物类似药声明标识
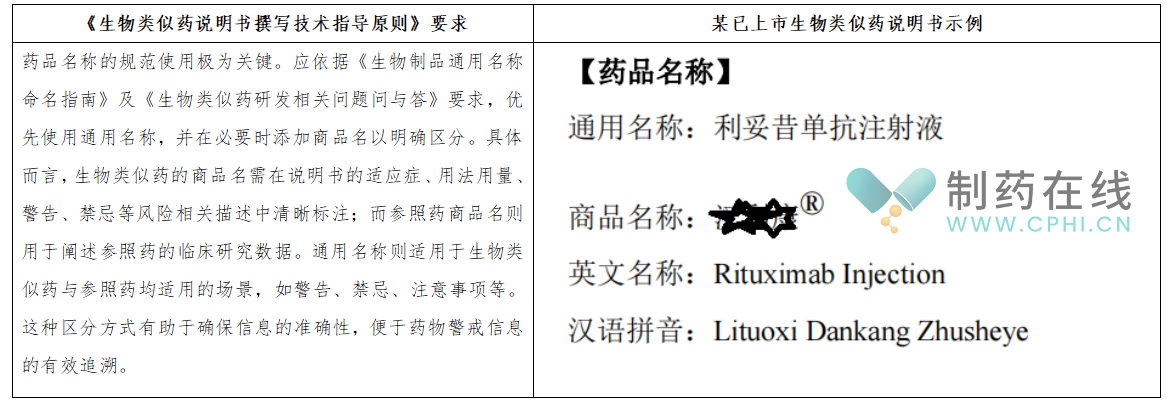
2.【药品名称】
3.【成份】

4.【适应症和用法用量】

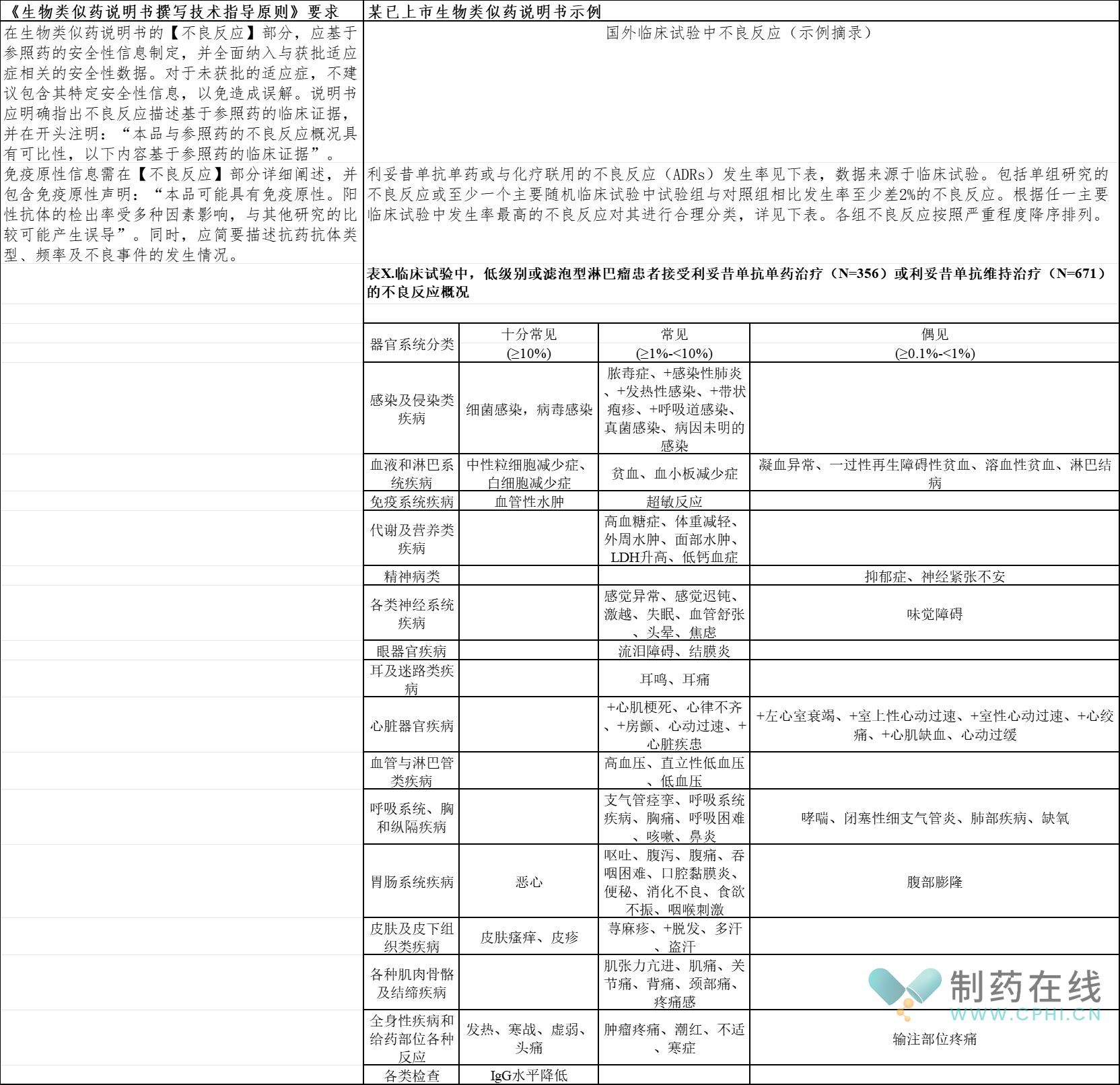
5.【不良反应】
6.【禁忌】

7.【注意事项】

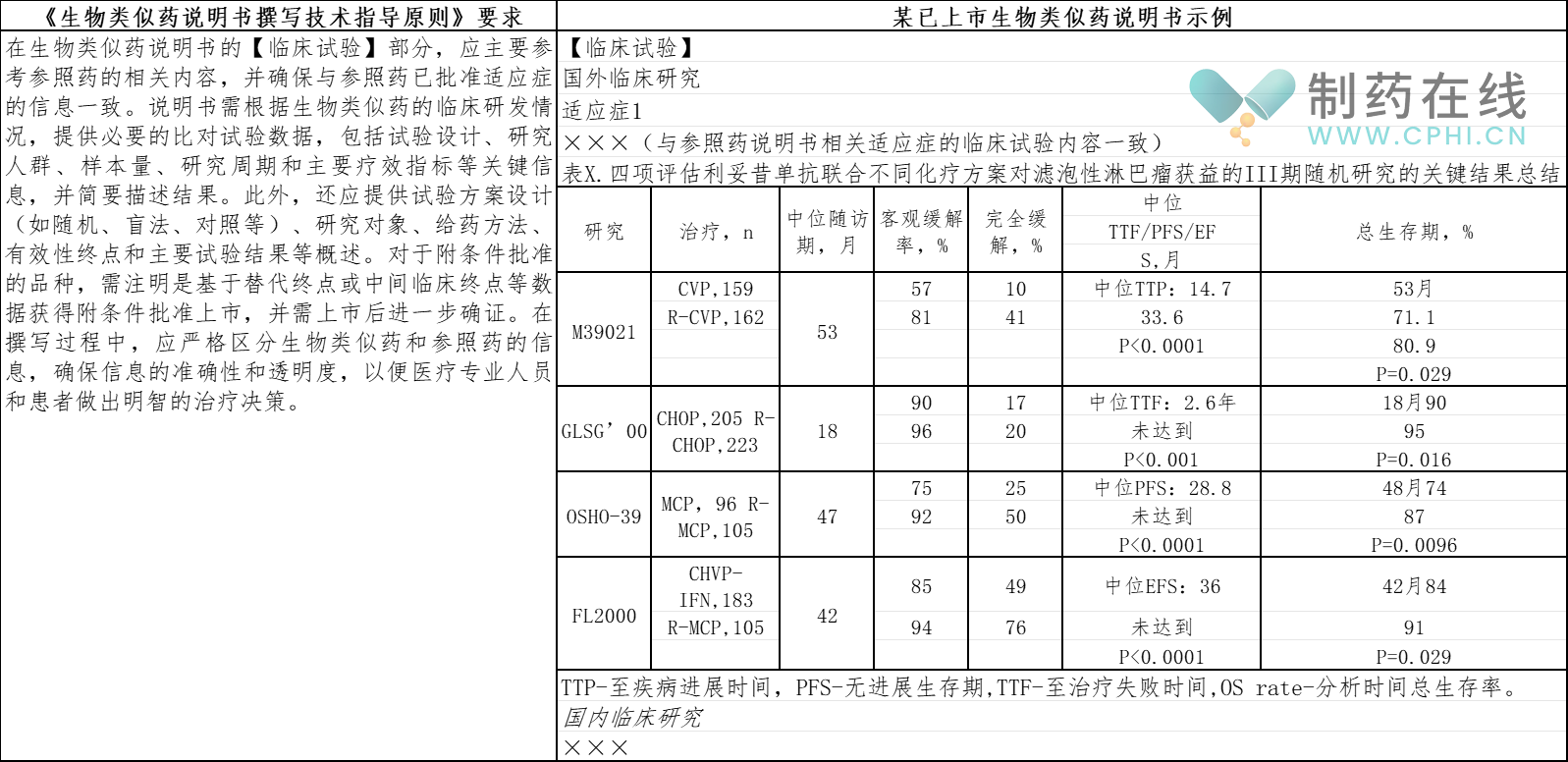
8.【临床试验】
9.【药理毒理】

参考文献:
[1]www.cde.org.cn、利妥昔单抗注射液说明书等



合作咨询
![]() 肖女士
肖女士
![]() 021-33392297
021-33392297
![]() Kelly.Xiao@imsinoexpo.com
Kelly.Xiao@imsinoexpo.com
 2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57
2006-2025 上海博华国际展览有限公司版权所有(保留一切权利)
沪ICP备05034851号-57






















